Akt/PKB signaling pathway
The Akt signaling pathway or PI3K-Akt signaling pathway is a signal transduction pathway that promotes survival and growth in response to extracellular signals. Key proteins involved are PI3K (phosphatidylinositol 3-kinase) and Akt (protein kinase B).
Initial stimulation by one of the growth factors causes activation of a cell surface receptor and phosphorylation of PI3K. Activated PI3K then phosphorylates lipids on the plasma membrane, forming second messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3). Akt, a serine/threonine kinase, is recruited to the membrane by interaction with these phosphoinositide docking sites, so that it can be fully activated.[1] Activated Akt mediates downstream responses, including cell survival, growth, proliferation, cell migration and angiogenesis, by phosphorylating a range of intracellular proteins. The pathway is present in all cells of higher eukaryotes and is highly conserved.[2]
The pathway is highly regulated by multiple mechanisms, often involving cross-talk with other signaling pathways. Problems with PI3K-Akt pathway regulation can lead to an increase in signaling activity. This has been linked to a range of diseases such as cancer and type 2 diabetes. A major antagonist of PI3K activity is PTEN (phosphatase and tensin homolog), a tumour suppressor which is often mutated or lost in cancer cells. Akt phosphorylates as many as 100 different substrates, leading to a wide range of effects on cells.[3]
Mechanism
PI3K activation
There are multiple types of phosphoinositide 3-kinase but only class I are responsible for lipid phosphorylation in response to growth stimuli. Class 1 PI3Ks are heterodimers composed of a regulatory subunit p85 and a catalytic subunit p110, named by their molecular weights.[4]
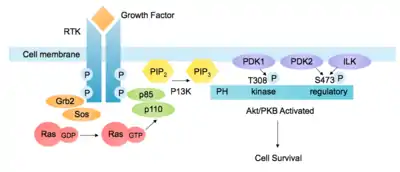
The pathway can be activated by a range of signals, including hormones, growth factors and components of the extracellular matrix (ECM).[5] It is stimulated by binding of an extracellular ligand to a receptor tyrosine kinase (RTK) in the plasma membrane, causing receptor dimerization and cross-phosphorylation of tyrosine residues in the intracellular domains. The regulatory subunit p85 binds to phosphorylated tyrosine residues on the activated receptor via its Src homology 2 (SH2) domain. It then recruits the catalytic subunit p110 to form the fully active PI3K enzyme. Alternatively, adaptor molecule Grb2 binds to phospho-YXN motifs of the RTK and recruits p85 via Grb2-associated binding (GAB) scaffold protein.[6]
The p110 subunit can also be recruited independently of p85. For example, Grb2 can also bind the Ras-GEF Sos1, leading to activation of Ras. Ras-GTP then activates the p110 subunit of PI3K. Other adaptor molecules such as insulin receptor substrate (IRS) can also activate p110. [7]
PI3K can also be activated by G protein-coupled receptors (GPCR), via G-protein βγ dimers or Ras which bind PI3K directly. In addition, the Gα subunit activates Src-dependent integrin signaling which can activate PI3K. [8]
Phosphoinositide formation
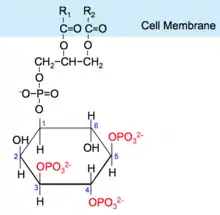
Activated PI3K catalyses the addition of phosphate groups to the 3'-OH position the inositol ring of phosphoinositides (PtdIns), producing three lipid products, PI(3)P, PI(3,4)P2 and PI(3,4,5)P3:
Phosphatidylinositol (PI) → PI 3-phosphate, (PI(4)P) → PI 3,4-bisphosphate, (PI(4,5)P2) → PI 3,4,5-triphosphate [9]
These phosphorylated lipids are anchored to the plasma membrane, where they can directly bind intracellular proteins containing a pleckstrin homology (PH) or FYVE domain. For example, the triphosphate form (PI(3,4,5)P3) binds Akt and phosphoinositide-dependent kinase 1 (PDK1) so they accumulate in close proximity at the membrane. [1][10]
Akt activation
Akt resides in the cytosol in an inactive conformation, until the cell is stimulated and it translocates to the plasma membrane. The Akt PH domain has a high affinity for second messenger PI(3,4,5)P3, binding to it preferentially over other phosphoinositides.[11] Thus PI3K activity is essential for translocation of Akt to the membrane. Interaction with PI(3,4,5)P3 causes conformational changes and exposure of phosphorylation sites Thr308 in the kinase domain and Ser473 in the C-terminal domain. Akt is partially activated by phosphorylation of T308 by PDK1. Full activation requires phosphorylation of S473, which can be catalysed by multiple proteins, including phosphoinositide-dependent kinase 2 (PDK2), integrin-linked kinase (ILK),[1] mechanistic target of rapamycin complex (mTORC) and DNA-dependent protein kinase (DNA-PK).[7][12] The regulation of Ser473 phosphorylation is not fully understood but may also be influenced by autophosphorylation after Thr308 phosphorylation. After stimulation, the levels of PIP3 decrease and Akt activity is attenuated by dephosphorylation by serine/threonine phosphatases.[5]
PI3K-independent activation
Although PI3K is the major mode of Akt activation, other tyrosine or serine/threonine kinases have been shown to activate Akt directly, in response to growth factors, inflammation or DNA damage. These can function even when PI3K activity is inhibited.[13] Other studies have shown Akt can be activated in response to heat shock[14] or increases in cellular Ca2+ concentration, via Ca2+/Calmodulin-dependent protein kinase kinase (CAMKK).[12][15]
| Activating Kinase | Akt Phosphorylation Site | Details |
|---|---|---|
| Activated CDC42 kinase 1 (Ack1) | Tyr176 | Akt binds preferentially to phosphatidic acid (PA) instead of PIP3 allowing translocation to the plasma membrane.[16] |
| Src | Tyr315, Tyr326 | Requires interaction of the Src SH3 domain and proline-rich region at the C-terminal of Akt.[17] |
| Protein tyrosine kinase 6 (PTK6) | Tyr215, Tyr315 and Tyr326 | Activates Akt in response to epidermal growth factor (EGF)[18] |
| IκB kinase ε (IKKε) | Ser137, Thr308 and Ser473 | Independent of the PH domain, PI3K, PDK1 and mTOR [19] |
| TANK-binding kinase 1 (TBK1) | Thr195, Ser378 and Ser473 | In response to Toll-like receptor activation in macrophages.[20] |
| DNA-dependent protein kinase (DNA-PK) | Ser473 | Activated by double-strand DNA breaks formed by ionizing radiation.[21] |
Regulation
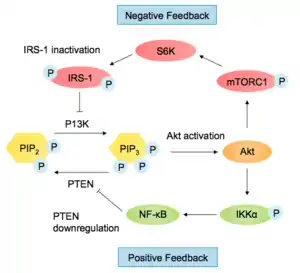
The PI3K-Akt pathway has many downstream effects and must be carefully regulated. One of the ways the pathway is negatively regulated is by reducing PIP3 levels. Phosphatase and tensin homolog (PTEN) antagonises PI3K by converting PI(3,4,5)P3 into PI(4,5)P2. Loss of PTEN function leads to over-activation of Akt and is common in cancer cells (PTEN is a tumour suppressor). SH2-containing Inositol Phosphatase (SHIP) also dephosphorylates PI(3,4,5)P3, at the 5' position of the inositol ring.[22] The PI3K-Akt pathway regulates PTEN levels by affecting its transcription and activity. Transcription factor NF-κB, activated by Akt, regulates peroxisome proliferator-activated receptor delta (PPARβ/δ) agonists and tumour necrosis factor α (TNFα), which in turn repress PTEN expression.[3] NEDD4-1, an E3 ligase that recognises PTEN for degradation is up-regulated by the PI3K pathway. Therefore, when Akt is activated, PTEN is further repressed in a positive feedback loop.[23]
The pathway is also controlled by protein phosphatase 2A (PP2A), which dephosphorylates Akt at Thr308 and phosphatase PHLPP dephosphorylates Akt at Ser473.[3] Another protein important in Akt attenuation is Carboxy Terminal Modulator Protein (CTMP). CTMP binds to the regulatory domain of Akt, blocking its phosphorylation and activation.[1]
When the pathway is activated by insulin, insulin receptor substrate 1 (IRS-1) transcription is down-regulated, in a negative feedback loop via mTORC1 and S6K1 activation. S6K1 is also able to phosphorylate IRS-1 at multiple serine residues, preventing binding to RTKs.[24] Another negative feedback control mechanism regulating the pathway involves FoxO transcription factors. Activated Akt causes FoxO degradation, so it can no longer inhibit PP2A, thus leading to a decrease in Akt phosphorylation.[3]
Downstream effects
Once active, Akt translocates from the plasma membrane to the cytosol and nucleus, where many of its substrates reside.[12] Akt regulates a wide range of proteins by phosphorylation. Akt target substrates contain a minimum consensus sequence R-X-R-X-X-[Ser/Thr]-Hyd, where Hyd is a hydrophobic amino acid, although other factors such as sub-cellular localisation and 3-dimensional structure are important.[5] Phosphorylation by Akt can be inhibitory or stimulatory, either suppressing or enhancing the activity of target proteins.
Cell survival and apoptosis
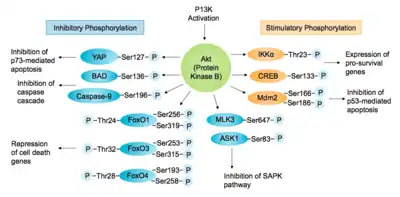
The Akt-PI3K pathway is essential for cell survival as activated Akt influences many factors involved in apoptosis, either by transcription regulation or direct phosphorylation. [5] In the nucleus, Akt inhibits transcription factors that promote the expression of cell death genes, and enhances transcription of anti-apoptotic genes. A well studied example is the Forkhead family transcription factors (FoxO/FH), of which FKHR/FoxO1, FKHRL1/FoxO3 and AFX/FoxO4 are directly phosphorylated by Akt.[12][25] This phosphorylation induces export to the cytosol where they are sequestered by 14-3-3 proteins and eventually undergo degradation via the ubiquitin-proteasome pathway.[2][26]
Akt also positively regulates some transcription factors to allow expression of pro-survival genes. Akt can phosphorylate and activate the IκB kinase IKKα, causing degradation of IκB and nuclear translocation of NF-κB where it promotes expression of caspase inhibitors, c-Myb and Bcl-xL.[2][12] Also promoting cell survival, cAMP response element binding protein (CREB) is phosphorylated by Akt at Ser133, stimulating recruitment of CREB-binding protein (CBP) to the promoter of target genes, such as Bcl-2.[27] Akt has also been shown to phosphorylate murine double minute 2 (Mdm2), a key regulator of DNA damage responses, at Ser166 and Ser186. Phosphorylation of Mdm2 by Akt upregulates its ubiquitin-ligase activity, therefore indirectly suppressing p53-mediated apoptosis.[25] Another target of Akt is the Yes-associated protein (YAP), phosphorylated at Ser127 leading to 14-3-3 binding and cytosolic localisation. Therefore, it cannot co-activate p73-mediated apoptosis in response to DNA damage.[28]
Akt negatively regulates pro-apoptotic proteins by direct phosphorylation. For example, phosphorylation of BAD, the Bcl-2 family member, on Ser136 causes translocation from the mitochondrial membrane to the cytosol, where it is sequestered by 14-3-3 proteins.[27] Akt phosphorylates Caspase-9 on Ser196, preventing a caspase cascade leading to cell death.[2][12] Akt also phosphorylates MAP kinase kinase kinases (MAPKKK) upstream of the stress-activated protein kinase (SAPK) pathway. Phosphorylation of apoptosis signal-regulating kinase 1 (ASK1) on Ser83 and mixed lineage kinase 3 (MLK3) on Ser674 inhibits their activity and prevents MAP kinase induced apoptosis.[25]
Lysosome biogenesis and autophagy
Akt regulates TFEB, a master controller of lysosomal biogenesis,[29] by direct phosphorylation of TFEB at serine 467.[30] Phosphorylated TFEB is excluded from the nucleus and less active.[30] Pharmacological inhibition of Akt promotes nuclear translocation of TFEB, lysosomal biogenesis and autophagy.[30]
Cell cycle progression

Akt promotes G1-S phase cell cycle progression by phosphorylating and inactivating glycogen synthase kinase 3 (GSK-3) at Ser9. This prevents the phosphorylation and degradation of cyclin D1. [31] Therefore, Akt promotes G1 phase progression in a positive feedback loop. Akt promotes cyclin D1 translation via indirect activation of mTOR. mTOR increases translation of cyclin D1 by activating ribosomal protein S6K, and inhibiting eukaryotic translation initiation factor 4E-binding protein (4E-BP), thus increasing eIF4e activity.[5][32]
Akt both indirectly and directly regulates cyclin-dependent kinase (CDK) inhibitors p21Cip1 and p27Kip1 , allowing cell cycle progression. Akt phosphorylates p27kip1 at Thr157, preventing its nuclear import.[33] In addition, Akt phosphorylates Thr145 and Ser146 of p21Cip1, preventing PCNA binding and decreasing stability.[34] Akt phosphorylation of Foxo transcription factors also affects the cell cycle, as inhibitory phosphorylation of FoxO4 (also named AFX) prevents p27 gene expression.[35]
Cell migration
Akt phosphorylates many proteins involved in polymerisation and stabilisation of the actin cytoskeleton. In normal cells, this can either increase the stability of cytoskeleton components or promote migration via remodelling. Examples are listed below:
- Actin filaments - Akt phosphorylates actin directly [36]
- Akt phosphorylation enhancer (APE), also named girdin - phosphorylated at Ser1416 causing translocation to the leading edge of filaments, essential for migration [37]
- Sodium-hydrogen exchanger 1 (NHE1) - phosphorylated at Ser648, promoting cytoskeletal rearrangements and migration [38]
- Filamin A - phosphorylated at Ser2152, promoting caveolin-1 mediated cell migration [39]
- Kank - kidney ankyrin repeat-containing protein - negatively regulating RhoA activation and cell migration in response to insulin and EGF [40]
- Tuberous sclerosis complex 2 (TSC2) - Akt1 destabilises the Rho GTPase, inhibits F-actin assembly and reduces cell migration [41]
- Palladin - Akt1 phosphorylates the actin-binding protein at Ser507, disrupting cross-linking of F-actin bundles [42]
Akt promotes cell migration by interacting with other cytoskeleton components. The type III intermediate filament Vimentin is phosphorylated by Akt1 at Ser39, preventing its degradation. In normal cells, this maintains tissue stability. S-phase kinase-associated protein 2 (Skp2) - Ser72 phosphorylation enhances E3 ligase activity and cytosolic localisation, promoting cell motility. Akt phosphorylates GSK3 beta, indirectly activating microtubule binding protein adenomatous polyposis coli (APC). Endothelial nitric oxide synthase (eNOS) is phosphorylated at Ser1177, leading to NO synthesis and endothelial cell migration.[43] In addition, the pro-migratory GTPase-activating protein RhoGAP22 is phosphorylated at Ser16.[36]
Oxidative stress
Under oxidative stress, miR-126 promotes Akt/PKB signaling pathway activation. This increases the biological function of cells under oxidative stress. This is important in endothelial progenitor cell transplantation to treat acute myocardial infarction (AMI) and may serve as a new therapeutic approach to treat AMI. [44]
Role in cancer
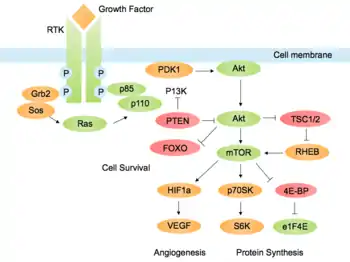
Aberrant activation of Akt, either via PI3K or independently of PI3K, is often associated with malignancy.[13] Studies have identified gene amplification of the Akt isoforms in many types of cancer, including glioblastoma, ovarian, pancreatic and breast cancers. Akt is also up-regulated in terms of mRNA production in breast and prostate cancer. Functional inactivation of PTEN, the major PI3K antagonist, can occur in cancer cells by point mutation, gene deletion or epigenetic mechanisms.[1] Mutation in the pathway can also affect receptor tyrosine kinases, growth factors, Ras and the PI3K p110 subunit, leading to abnormal signaling activity. Therefore, many of the proteins in the pathway are targets for cancer therapeutics.[45] In addition to its effects on cell survival and cell cycle progression, the PI3K-Akt pathway promotes other characteristics of cancer cells. Hyperactivity of the pathway promotes the epithelial-mesenchymal transition (EMT) and metastasis due to its effects on cell migration.[36]
Angiogenesis
Angiogenesis, the formation of new blood vessels, is often critical for tumour cells to survive and grow in nutrient-depleted conditions. Akt is activated downstream of vascular endothelial growth factor (VEGF) in endothelial cells in the lining of blood vessels, promoting survival and growth. Akt also contributes to angiogenesis by activating endothelial nitric oxide synthase (eNOS), which increases production of nitric oxide (NO). This stimulates vasodilation and vascular remodelling.[2] Signaling through the PI3K-Akt pathway increases translation of hypoxia-inducible factor α (HIF1α and HIF2α) transcription factors via mTOR.[46] HIF promotes gene expression of VEGF and glycolytic enzymes, allowing metabolism in oxygen-depleted environments.[47]
Glucose metabolism
In cancer cells, an increase in Akt signaling correlates with an increase in glucose metabolism, compared to normal cells. Cancer cells favour glycolysis for energy production over mitochondrial oxidative phosphorylation, even when oxygen supply is not limited. This is known as the Warburg effect, or aerobic glycolysis. Akt affects glucose metabolism by increasing translocation of glucose transporters GLUT1 and GLUT4 to the plasma membrane, increasing hexokinase expression and phosphorylating GSK3 which stimulates glycogen synthesis.[5] It also activates glycolysis enzymes indirectly, via HIF transcription factors and phosphorylation of phosphofructokinase-2 (PFK2) which activates phosphofructokinase-1 (PFK1).[48]
References
- Osaki M, Oshimura M, Ito H (2004). "The PI3K-Akt pathway: Its functions and alterations in human cancer". Apoptosis. 9 (6): 667–676. doi:10.1023/B:APPT.0000045801.15585.dd. PMID 15505410.
- Manning BD, Cantley LC (2007). "AKT/PKB Signaling: Navigating Downstream". Cell. 129 (7): 1261–1274. doi:10.1016/j.cell.2007.06.009. PMC 2756685. PMID 17604717.
- Carracedo A, Pandolfi PP (2008). "The PTEN-PI3K pathway: of feedbacks and cross-talks". Oncogene. 27 (41): 5527–5541. doi:10.1038/onc.2008.247. PMID 18794886.
- Cantrell DA (2001). "Phosphoinositide 3-kinase signalling pathways". J Cell Sci. 114 (Pt 8): 1439–1445. PMID 11282020.
- Nicholson KM, Anderson NG (2002). "The protein kinase B/Akt signalling pathway in human malignancy". Cellular Signalling. 14 (5): 381–395. doi:10.1016/S0898-6568(01)00271-6. PMID 11882383.
- Castellano E, Downward J (2011). "RAS Interaction with PI3K". Genes Cancer. 2 (3): 261–74. doi:10.1177/1947601911408079. PMC 3128635. PMID 21779497.
- Hemmings BA, Restuccia DF (2012). "PI3K-PKB/Akt Pathway". Cold Spring Harb Perspect Biol. 4 (9): a011189. doi:10.1101/cshperspect.a011189. PMC 3428770. PMID 22952397.
- New DC, Wong YH (2007). "Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression". J Mol Signall. 2 (2): 2. doi:10.1186/1750-2187-2-2. PMC 1808056. PMID 17319972.
- Fruman DA, Meyers RE, Cantley LC (1998). "Phosphoinositide Kinases". Annu Rev Biochem. 67: 481–507. doi:10.1146/annurev.biochem.67.1.481. PMID 9759495.
- Cantley LC (2002). "The Phosphoinositide 3-Kinase Pathway". Science. 296 (5573): 1655–1657. doi:10.1126/science.296.5573.1655. PMID 12040186.
- Miao B, Skidan I, Yang J, Lugovskoy A, Reibarkh M, Long K, Brazell T, Durugkar KA, Maki J, Ramana CV, Schaffhausen B, Wagner G, Torchilin V, Yuan J, Degterev A (2010). "Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains". Proc. Natl. Acad. Sci. USA. 107 (46): 20126–20131. doi:10.1073/pnas.1004522107. PMC 2993381. PMID 21041639.
- Vanhaesebroeck B, Alessi DR (2000). "The PI3K-PDK1 connection: more than just a road to PKB". Biochem J. 346 (3): 561–576. doi:10.1042/0264-6021:3460561. PMC 1220886. PMID 10698680.
- Mahajan K, Mahajam NP (2012). "PI3K-Independent AKT activation in Cancers: A Treasure Trove for Novel Therapeutics". J Cell Physiol. 227 (9): 3178–3184. doi:10.1002/jcp.24065. PMC 3358464. PMID 22307544.
- M Shaw; P Cohen; D R Alessi (1998). "The activation of protein kinase B by H2O2 or heat shock is mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated protein kinase-2". Biochem J. 336 (1): 241–246. doi:10.1042/bj3360241. PMC 1219864. PMID 9806907.
- Soderling TR (1999). "The Ca-calmodulin-dependent protein kinase cascade". Trends Biochem. Sci. 24 (6): 232–236. doi:10.1016/S0968-0004(99)01383-3. PMID 10366852.
- Mahajan K, Mahajam NP (2010). "Shepherding AKT and androgen receptor by Ack1 tyrosine kinase". J Cell Physiol. 224 (2): 327–33. doi:10.1002/jcp.22162. PMC 3953130. PMID 20432460.
- Jiang T1 & Qiu Y (2003). "Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation". J Biol Chem. 278 (18): 15789–93. doi:10.1074/jbc.M212525200. PMID 12600984.
- Zheng Y, Peng M, Wang Z, Asara JM, Tyner AL (2010). "Protein tyrosine kinase 6 directly phosphorylates AKT and promotes AKT activation in response to epidermal growth factor". Mol Cell Biol. 30 (17): 4280–92. doi:10.1128/MCB.00024-10. PMC 2937549. PMID 20606012.
- Guo JP, Coppola D, Cheng JQ (2011). "IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation". J Biol Chem. 286 (43): 37389–98. doi:10.1074/jbc.M111.287433. PMC 3199486. PMID 21908616.
- Joung SM, Park ZY, Rani S, Takeuchi O, Akira S, Lee JY (2011). "Akt contributes to activation of the TRIF-dependent signaling pathways of TLRs by interacting with TANK-binding kinase 1". J Immunol. 186 (1): 499–507. doi:10.4049/jimmunol.0903534. PMID 21106850.
- Toulany M, Rodemann HP (2013). "Potential of Akt mediated DNA repair in radioresistance of solid tumors overexpressing erbB-PI3K-Akt pathway". Translational Cancer Research. 2 (3). doi:10.3978/j.issn.2218-676X.2013.04.09.
- Georgescu MM (2010). "PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control". Genes Cancer. 1 (12): 1170–1177. doi:10.1177/1947601911407325. PMC 3092286. PMID 21779440.
- Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, Pandolfi PP, Jiang X (2007). "NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN". Cell. 128 (1): 129–139. doi:10.1016/j.cell.2006.11.039. PMC 1828909. PMID 17218260.
- Zhang J, Gao Z, Yin J, Quon MJ, Ye J (2008). "S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2". J Biol Chem. 283 (51): 35375–82. doi:10.1074/jbc.M806480200. PMC 2602883. PMID 18952604.
- Song G, Ouyang G, Bao S (2005). "The activation of Akt/PKB signalling pathway and cell survival". J Cell Mol Med. 9 (1): 59–71. doi:10.1111/j.1582-4934.2005.tb00337.x. PMC 6741304. PMID 15784165.
- Zhang X, Tang N, Hadden TJ, Rishi AK (2011). "Akt, FoxO and regulation of apoptosis". Biochim Biophys Acta. 1813 (11): 1978–1986. doi:10.1016/j.bbamcr.2011.03.010. PMID 21440011.
- Du K, Montminy M (1998). "CREB Is a Regulatory Target for the Protein Kinase Akt/PKB". J Biol Chem. 273 (49): 32377–32379. doi:10.1074/jbc.273.49.32377. PMID 9829964.
- Basu S, Totty NF, Irwin MS, Sudol M, Downward J (2003). "Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis". Mol Cell. 11 (1): 11–23. doi:10.1016/S1097-2765(02)00776-1. PMID 12535517.
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (Jul 2009). "A gene network regulating lysosomal biogenesis and function". Science. 325 (5939): 473–7. doi:10.1126/science.1174447. PMID 19556463.
- Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, Chaudhury A, Bajaj L, Bondar VV, Bremner L, Saleem U, Tse DY, Sanagasetti D, Wu SM, Neilson JR, Pereira FA, Pautler RG, Rodney GG, Cooper JD, Sardiello M (Feb 2017). "mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases". Nature Communications. 8: 14338. doi:10.1038/ncomms14338. PMC 5303831. PMID 28165011.
- Alao JP (2007). "The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention". Molecular Cancer. 6 (24): 24. doi:10.1186/1476-4598-6-24. PMC 1851974. PMID 17407548.
- Hay N, Sonenberg N (2004). "Upstream and downstream of mTOR". Genes Dev. 18 (16): 1926–45. doi:10.1101/gad.1212704. PMID 15314020.
- Denicourt C, Dowdy S (2004). "Cip/Kip proteins: more than just CDKs inhibitors". Genes Dev. 18 (8): 851–855. doi:10.1101/gad.1205304. PMID 15107401.
- Li Y, Dowbenko D, Lasky LA (2001). "AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival". J Biol Chem. 277 (13): 11352–11361. doi:10.1074/jbc.M109062200. PMID 11756412.
- Liang J, Slingerland JM (2003). "Multiple Roles of the PI3K/PKB (Akt) Pathway in Cell Cycle Progression". Cell Cycle. 2 (4): 336–342. doi:10.4161/cc.2.4.433. PMID 12851486.
- Xue G, Hemmings BA (2013). "PKB/Akt-Dependent Regulation of Cell Motility". J Natl Cancer Inst. 105 (6): 393–404. doi:10.1093/jnci/djs648. PMID 23355761.
- Enomoto A, Ping J, Takahashi M (2006). "Girdin, a novel actin-binding protein, and its family of proteins possess versatile functions in the Akt and Wnt signaling pathways". Ann N Y Acad Sci. 1086: 169–184. doi:10.1196/annals.1377.016. PMID 17185515.
- Meima ME, Webb BA, Witkowska HE, Barber DL (2009). "The Sodium-Hydrogen Exchanger NHE1 Is an Akt Substrate Necessary for Actin Filament Reorganization by Growth Factors". J Biol Chem. 284 (39): 26666–26675. doi:10.1074/jbc.M109.019448. PMC 2785354. PMID 19622752.
- Ravid D, Chuderland D, Landsman L, Lavie Y, Reich R, Liscovitch M (2008). "Filamin A is a novel caveolin-1-dependent target in IGF-I-stimulated cancer cell migration". Exp Cell Res. 314 (15): 2762–2773. doi:10.1016/j.yexcr.2008.06.004. PMID 18598695.
- Kakinuma N, Roy BC, Zhu Y, Wang Y, Kiyama R (2008). "Kank regulates RhoA-dependent formation of actin stress fibers and cell migration via 14-3-3 in PI3K–Akt signaling". J Cell Biol. 181 (3): 537–549. doi:10.1083/jcb.200707022. PMC 2364698. PMID 18458160.
- Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002). "TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling". Nat Cell Biol. 4 (9): 648–657. doi:10.1038/ncb839. PMID 12172553.
- Chin YR, Toker A (2010). "Akt2 regulates expression of the actin-bundling protein palladin". FEBS Lett. 584 (23): 4769–4774. doi:10.1016/j.febslet.2010.10.056. PMC 2997733. PMID 21050850.
- Lamalice L, Le Boeuf F, Huot J (2007). "Endothelial Cell Migration During Angiogenesis". Circulation Research. 100 (6): 782–794. doi:10.1161/01.RES.0000259593.07661.1e. PMID 17395884.
- Wu Q, Qi B, Duan X, Ming X, Yan F, He Y, Bu X, Sun S, Zhu H. MicroRNA-126 enhances the biological function of endothelial progenitor cells under oxidative stress via PI3K/Akt/GSK-3β and ERK1/2 signaling pathways. Bosn J of Basic Med Sci [Internet]. 2020Jan.30 [cited 2020Mar.14];. Available from: https://www.bjbms.org/ojs/index.php/bjbms/article/view/4493
- Yuan TL Cantley LC (2008). "PI3K pathway alterations in cancer: variations on a theme". Oncogene. 27 (41): 5497–5510. doi:10.1038/onc.2008.245. PMC 3398461. PMID 18794884.
- Karar J, Maity A (2011). "PI3K/AKT/mTOR Pathway in Angiogenesis". Front Mol Neurosci. 4 (51): 51. doi:10.3389/fnmol.2011.00051. PMC 3228996. PMID 22144946.
- Liu W, Shen SM, Zhao XY, Chen GQ (2012). "Targeted genes and interacting proteins of hypoxia inducible factor-1". Int J Biochem Mol Biol. 3 (2): 165–178. PMC 3388736. PMID 22773957.
- Simons, Andrean L; Orcutt, Kevin P; Madsen, Joshua M; Scarbrough, Peter M; Spitz, Douglas R (2012). Oxidative Stress in Cancer Biology and Therapy. Humana Press. pp. 21–46. ISBN 978-1617793967.