Aromatic ring current
An aromatic ring current is an effect observed in aromatic molecules such as benzene and naphthalene. If a magnetic field is directed perpendicular to the plane of the aromatic system, a ring current is induced in the delocalized π electrons of the aromatic ring.[1] This is a direct consequence of Ampère's law; since the electrons involved are free to circulate, rather than being localized in bonds as they would be in most non-aromatic molecules, they respond much more strongly to the magnetic field.
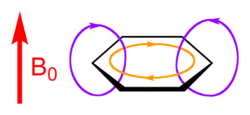
The ring current creates its own magnetic field. Outside the ring, this field is in the same direction as the externally applied magnetic field; inside the ring, the field counteracts the externally applied field. As a result, the net magnetic field outside the ring is greater than the externally applied field alone, and is less inside the ring.
Aromatic ring currents are relevant to NMR spectroscopy, as they dramatically influence the chemical shifts of 1H nuclei in aromatic molecules.[2] The effect helps distinguish these nuclear environments and is therefore of great use in molecular structure determination. In benzene, the ring protons experience deshielding because the induced magnetic field has the same direction outside the ring as the external field and their chemical shift is 7.3 ppm compared to 5.6 for the vinylic proton in cyclohexene. In contrast any proton inside the aromatic ring experiences shielding because both fields are in opposite direction. This effect can be observed in cyclooctadecanonaene ([18]annulene) with 6 inner protons at −3 ppm.
The situation is reversed in antiaromatic compounds. In the dianion of [18]annulene the inner protons are strongly deshielded at 20.8 ppm and 29.5 ppm with the outer protons significantly shielded (with respect to the reference) at −1.1 ppm. Hence a diamagnetic ring current or diatropic ring current is associated with aromaticity whereas a paratropic ring current signals antiaromaticity.
A similar effect is observed in three-dimensional fullerenes; in this case it is called a sphere current.[3]
Relative aromaticity
Numerous attempts have been made to quantify aromaticity with respect to the observed ring current.[4] One method is called diamagnetic susceptibility exaltation Λ defined as the difference between the measured magnetic susceptibility of a compound and a calculated value based on group additivity tables. Large negative values are aromatic, for example, benzene (Λ = −13.4). Values close to zero are non-aromatic, for example, borazine (Λ = −1.7) and cyclohexane (Λ = 1.1). Large positive values are antiaromatic, for example, cyclobutadiene (Λ = +18).
Another measurable quantity is the chemical shift of lithium ions Li+ in complexes of lithium with aromatic structures because lithium tends to bond as a π-coordinate complex to the face of the aromatic rings. Thus the lithium atom in cyclopentadienyl lithium (CpLi) has a chemical shift of −8.6 ppm (aromatic) and its Cp2Li− complex a shift of −13.1.
Both methods suffer from the disadvantage that values depend on ring size.
| Chemical | ppm |
|---|---|
| Pyrrole | −15.1 |
| Thiophene | −13.6 |
| Furan | −12.3 |
| naphthalene | −9.9 |
| Benzene | −9.7 |
| Tropylium | −7.6 |
| Cyclopentadiene | −3.2 |
| Cyclohexane | −2.2 |
| Pentalene | 18.1 |
| Heptalene | 22.7 |
| Cyclobutadiene | 27.6 |
Nucleus-independent chemical shift
The nucleus-independent chemical shift (NICS) is a computational method that calculates the absolute magnetic shielding at the center of a ring. The values are reported with a reversed sign to make them compatible with the chemical shift conventions of NMR spectroscopy.[5] In this method, negative NICS values indicate aromaticity and positive values antiaromaticity.
Harmonic oscillator model of aromaticity
Yet another method called the harmonic oscillator model of aromaticity (HOMA)[6] is defined as a normalized sum of squared deviations of bond lengths from the optimal value, which is assumed to be realized for a fully aromatic system.[7] An aromatic compound has HOMA value 1 whereas a non-aromatic compound has value 0. For all-carbon systems, the HOMA value is defined as:

where 257.7 the normalization value, n is the number of carbon–carbon bonds, and d are bond lengths in angstrom (dopt is optimized (1.388 Å) and di is the experimental or computed).
References
- The induced magnetic field in cyclic molecules. Merino, G.; Heine, T.; Seifert, G. Chem. Eur. J.; 2004; 10; 4367-4371. doi:10.1002/chem.200400457
- Aromaticity and Ring Currents. Gomes, J. A. N. F.; Mallion, R. B. Chem. Rev.; (Review); 2001; 101(5); 1349-1384. doi:10.1021/cr990323h
- Sphere Currents of Buckminsterfullerene, Mikael P. Johansson, Jonas Jusélius, and Dage Sundholm, Angew. Chem. Int. Ed., Vol. 44, No. 12, pp. 1843-1846, 2005 doi:10.1002/anie.200462348 PMID 15706578
- What is aromaticity? Paul von Ragué Schleyer and Haijun Jiao Pure Appl. Chem., Vol. 68, No. 2, pp. 209-218, 1996 Link
- Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe Paul von Ragué Schleyer, Christoph Maerker, Alk Dransfeld, Haijun Jiao, and Nicolaas J. R. van Eikema Hommes J. Am. Chem. Soc.; 1996; 118(26) pp 6317-6318; (Communication) doi:10.1021/ja960582d
- Definition of aromaticity basing on the harmonic oscillator model Tetrahedron Letters, Volume 13, Issue 36, 1972, Pages 3839-3842 J. Kruszewski and T. M. Krygowski doi:10.1016/S0040-4039(01)94175-9
- How far is the π-electron delocalization of the phenanthrene moiety modified in the aza-analogues and their N-oxides? Beata T. Stępień, Tadeusz M. Krygowski,a Michał K. Cyrański, Jacek Młochowski, Pierluigi Orioli, and Francesco Abbate Arkivoc 2003 Link