ChIP-exo
ChIP-exo is a chromatin immunoprecipitation based method for mapping the locations at which a protein of interest (transcription factor) binds to the genome. It is a modification of the ChIP-seq protocol, improving the resolution of binding sites from hundreds of base pairs to almost one base pair. It employs the use of exonucleases to degrade strands of the protein-bound DNA in the 5'-3' direction to within a small number of nucleotides of the protein binding site. The nucleotides of the exonuclease-treated ends are determined using some combination of DNA sequencing, microarrays, and PCR. These sequences are then mapped to the genome to identify the locations on the genome at which the protein binds.
Theory
Chromatin immunoprecipitation (ChIP) techniques have been in use since 1984[1] to detect protein-DNA interactions. There have been many variations on ChIP to improve the quality of results. One such improvement, ChIP-on-chip (ChIP-chip), combines ChIP with microarray technology. This technique has limited sensitivity and specificity, especially in vivo where microarrays are constrained by thousands of proteins present in the nuclear compartment, resulting in a high rate of false positives.[2] Next came ChIP-sequencing (ChIP-seq), which combines ChIP with high-throughput sequencing.[3] However, the heterogeneous nature of sheared DNA fragments maps binding sites to within ±300 base pairs, limiting specificity. Secondly, contaminating DNA presents a grave problem since so few genetic loci are cross-linked to the protein of interest, making any non-specific genomic DNA a significant source of background noise.[4]
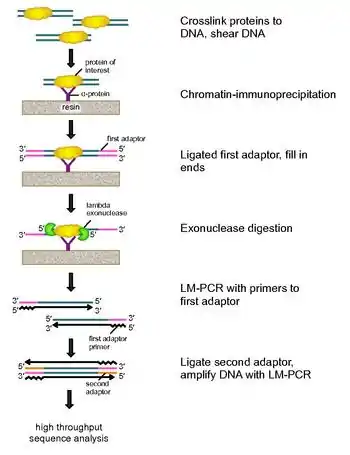
To address these problems, Rhee and Pugh revised the classic nuclease protection assay to develop ChIP-exo.[5] This new ChIP technique relies on a lambda exonuclease that degrades only, and all, unbound double-stranded DNA in the 5′-3′ direction. Briefly, a protein of interest (engineering one with an epitope tag can be useful for immunoprecipitation) is crosslinked in vivo to its natural binding locations across a genome using formaldehyde. Cells are then collected, broken open, and the chromatin sheared and solubilized by sonication. An antibody is then used to immunoprecipitate the protein of interest, along with the crosslinked DNA. DNA PCR adaptors are then ligated to the ends, which serve as a priming point for second strand DNA synthesis after the exonuclease digestion. Lambda exonuclease then digests double DNA strands from the 5′ end until digestion is blocked at the border of the protein-DNA covalent interaction. Most contaminating DNA is degraded by the addition of a second single-strand specific exonuclease. After the cross-linking is reversed, the primers to the PCR adaptors are extended to form double stranded DNA, and a second adaptor is ligated to 5′ ends to demarcate the precise location of exonuclease digestion cessation. The library is then amplified by PCR, and the products are identified by high throughput sequencing. This method allows for resolution of up to a single base pair for any protein binding site within any genome, which is a much higher resolution than either ChIP-chip or ChIP-seq.
Advantages
ChIP-exo has been shown to give up to single base pair resolution in identifying protein binding locations. This is in contrast to ChIP-seq which can locate a protein's binding site only to with ±300 base pairs.[4]
Contamination of non-protein-bound DNA fragments can result in a high rate of false positives and negatives in ChIP experiments. The addition of exonucleases to the process not only improves resolution of binding-site calling, but removes contaminating DNA from the solution before sequencing.[4]
Proteins that are inefficiently bound to a nucleotide fragment are more likely to be detected by ChIP-exo. This has allowed, for example, the recognition of more CTCF transcription factor binding sites than previously discovered.[5]
Due to the higher resolution and reduced background, less depth of sequencing coverage is needed when using ChIP-exo.[4]
Limitations
If a protein-DNA complex has multiple locations of cross-linking within a single binding event, then it can appear as though there are multiple distinct binding events. This likely results from these proteins being denatured and cross-linking at one of the available binding sites within the same event. The exonuclease would then stop at one of the bound sites, depending on which site the protein is cross-linked to.[5]
As with any ChIP-based method, a suitable antibody for the protein of interest needs to be available in order to use this technique.
Applications
Rhee and Pugh introduce ChIP-exo by performing analyses on a small collection of transcription factors: Reb1, Gal4, Phd1, Rap1 in yeast and CTCF in human. Reb1 sites were often found in clusters and these clusters had ~10-fold higher occupancy than expected. Secondary sites in clusters were found ~40 bp from a primary binding site. Binding motifs of Gal4 showed a strong preference for three of the four nucleotides, suggesting a negative interaction between Gal4 and the excluded nucleotide. Phd1 recognizes three different motifs which explains previous reports of the ambiguity of Phd1's binding motif. Rap1 was found to recognize four motifs. Ribosomal protein genes bound by this protein had a tendency to use a particular motif with a stronger consensus sequence. Other genes often used clusters of weaker consensus motifs, possibly to achieve a similar occupancy. Binding motifs of CTCF employed four "modules". Half of the bound CTCF sites used modules 1 and 2, while the rest used some combination of the four. It is believed that CTCF uses its zinc fingers to recognize different combinations of these modules.[5]
Rhee and Pugh analyzed pre-initiation complex (PIC) structure and organization in Saccharomyces genomes. Using ChIP-exo, they were able to, among other discoveries, precisely identify TATA-like features in promoters reported to be TATA-less.[6]
References
- Gilmour, DS; JT Lis (1983). "Detecting protein-DNA interactions in vivo: Distribution of RNA polymerase on specific bacterial genes". Proceedings of the National Academy of Sciences. 81 (14): 4275–4279. doi:10.1073/pnas.81.14.4275. PMC 345570. PMID 6379641.
- Albert, I; TN Mavrich; LP Tomsho; J Qi; SJ Zanton; SC Schuster; BF Pugh (2007). "Translational and rotational settings of H2A.Z nucleosomes cross the Saccharomyces cerevisiae genome". Nature. 446 (7135): 572–576. Bibcode:2007Natur.446..572A. doi:10.1038/nature05632. PMID 17392789. S2CID 4416890.
- Ren, B; F Robert; JJ Wyrick; O Aparicio; EG Jennings; I Simon; J Zeitlinger; J Schreiber; N Hannett; E Kan; et al. (2000). "Genome-wide location and function of DNA binding proteins". Science. 290 (5500): 2306–2309. Bibcode:2000Sci...290.2306R. CiteSeerX 10.1.1.123.6772. doi:10.1126/science.290.5500.2306. PMID 11125145.
- Pugh, Benjamin. "Methods, Systems and Kits for Detecting Protein-Nucleic Acid Interactions". United States Application Publication. United States Patents. Retrieved 17 February 2012.
- Rhee, Ho Sung; BJ Pugh (2011). "Comprehensive Genome-wide Protein-DNA Interactions Detected at Single-Nucleotide Resolution". Cell. 147 (6): 1408–1419. doi:10.1016/j.cell.2011.11.013. PMC 3243364. PMID 22153082.
- Rhee, Ho Sung; BJ Pugh (2012). "Genome-wide structure and organization of eukaryotic pre-initiation complexes". Nature. 483 (7389): 295–301. Bibcode:2012Natur.483..295R. doi:10.1038/nature10799. PMC 3306527. PMID 22258509.
External links
- DNA-protein interactions in high definition
- Resolving transcription factor binding
- High-resolution chromatin immunoprecipitation
- Important Gene-Regulation Proteins Pinpointed by New Method
- CexoR: An R/Bioconductor Package to Uncover High-resolution Protein-DNA Interactions in ChIP-exo Replicates
- Peconic Genomics