Chemical glycosylation
A chemical glycosylation reaction involves the coupling of a glycosyl donor, to a glycosyl acceptor forming a glycoside.[1][2][3] If both the donor and acceptor are sugars, then the product is an oligosaccharide. The reaction requires activation with a suitable activating reagent. The reactions often result in a mixture of products due to the creation of a new stereogenic centre at the anomeric position of the glycosyl donor. The formation of a glycosidic linkage allows for the synthesis of complex polysaccharides which may play important roles in biological processes and pathogenesis and therefore having synthetic analogs of these molecules allows for further studies with respect to their biological importance.
Terminology
The glycosylation reaction involves the coupling of a glycosyl donor and a glycosyl acceptor via initiation using an activator under suitable reaction conditions.
- A glycosyl donor is a sugar with a suitable leaving group at the anomeric position. This group, under the reaction conditions, is activated and via the formation of an oxocarbenium is eliminated leaving an electrophilic anomeric carbon.
- A glycosyl acceptor is a sugar with an unprotected nucleophilic hydroxyl group which may attack the carbon of the oxocarbenium ion formed during the reaction and allow for the formation of the glycosidic bond.
An activator is commonly a Lewis acid which enables the leaving group at the anomeric position to leave and results in the formation of the oxocarbenium ion.
Stereochemistry
The formation of a glycosidic linkage results in the formation of a new stereogenic centre and therefore a mixture of products may be expected to result. The linkage formed may either be axial or equatorial (α or β with respect to glucose). To better understand this, the mechanism of a glycosylation reaction must be considered.
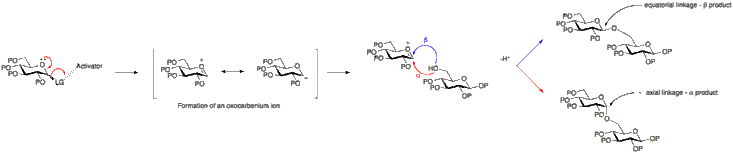
Neighbouring group participation
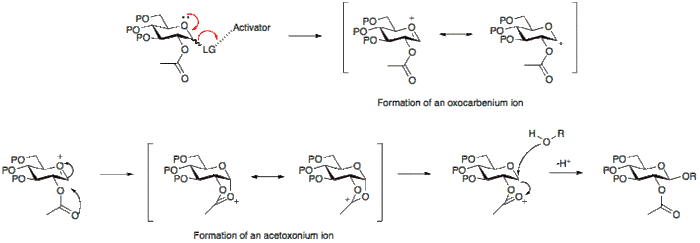
The stereochemical outcome of a glycosylation reaction may in certain cases be affected by the type of protecting group employed at position 2 of the glycosyl donor. A participating group, typically one with a carboxyl group present, will predominantly result in the formation of a β-glycoside. Whereas a non-participating group, a group usually without a carboxyl group, will often result in an α-glycoside.
Below it can be seen that having an acetyl protecting group at position 2 allows for the formation for an acetoxonium ion intermediate that blocks attack to the bottom face of the ring therefore allowing for the formation of the β-glycoside predominantly.
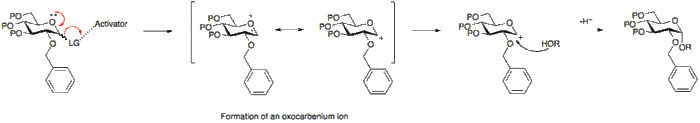
Alternatively, the absence of a participating group at position 2, allows for either attack from the bottom or top face. Since the α-glycoside product will be favoured by the anomeric effect, the α-glycoside usually predominates.
Protecting groups
Different protecting groups on either the glycosyl donor or the glycosyl acceptor[4][5] may affect the reactivity and yield of the glycosylation reaction. Typically, electron-withdrawing groups such as acetyl or benzoyl groups are found to decrease the reactivity of the donor/acceptor and are therefore termed "disarming" groups. Electron-donating groups such as the benzyl group, are found to increase the reactivity of the donor/acceptor and are therefore called "arming" groups.
Current methods in glycoside synthesis
Glycosyl iodides
Glycosyl iodides were first introduced for use in glycosylation reactions in 1901 by Koenigs and Knorr[6][7] although were often considered too reactive for synthetic use. Recently several research groups have shown these donors to have unique reactive properties and can differ from other glycosyl chlorides or bromides with respect to reaction time, efficiency, and stereochemistry.[8][9][10][11] Glycosyl iodides may be made under a variety of conditions, one method of note is the reaction of a 1-O-acetylpyranoside with TMSI.[12]

Iodide donors may typically be activated under basic conditions to give β-glycosides with good selectivity. The use of tetraalkylammonium iodide salts such as tetrabutylammonium iodide (TBAI) allows for in-situ anomerization of the α-glycosyl halide to the β-glycosyl halide and provides the α-glycoside in good selectivity.[13][14][15][16]

Thioglycosides
Thioglycosides were first reported in 1909 by Fischer [17] and since then have been explored constantly allowing for the development of numerous protocols for their preparation. The advantage of using thioglycosides is their stability under a wide range of reaction conditions allowing for protecting group manipulations. Additionally thioglycosides act as temporary protecting groups at the anomeric position allowing for thioglycosides to be useful as both glycosyl donors as well as glycosyl acceptors.[13] Thioglycosides are usually prepared by reacting per-acetylated sugars with BF3•OEt2 and the appropriate thiol.[18][19][20]
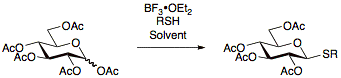
Thioglycosides used in glycosylation reactions as donors can be activated under a wide range of conditions, most notably using NIS/AgOTf.[21]
Trichloroacetimidates
Trichloroacetimidates were first introduced and explored by Schmidt in 1980[22][23] and since then have become very popular for glycoside synthesis. The use of trichloroacetimidates provides many advantages including ease of formation, reactivity and stereochemical outcome.[13] O-Glycosyl trichloroacetimidates are prepared via the addition of trichloroacetonitrile (Cl3CCN) under basic conditions to a free anomeric hydroxyl group.
Typical activating groups for glycosylation reactions using trichloroacetimidates are BF3•OEt2 or TMSOTf.[24]
Column chromatographic purification of the reaction mixture can sometimes be challenging due to the trichloroacetamide by-product. This can, however, be overcome by washing the organic layer with 1 M NaOH solution in a separatory funnel prior to chromatography. Acetyl protecting groups were found to be stable during this procedure.[25]
Notable synthetic products
Below are a few examples of some notable targets obtained via a series of glycosylation reactions.

See also
References
- Boons, Geert-Jan; Karl J. Hale (2000). Organic synthesis with carbohydrates. Blackwell Publishing. ISBN 978-1-85075-913-3.
- Crich, D.; Lim, L. Org. React. 2004, 64, 115. doi:10.1002/0471264180.or064.02
- Bufali, S.; Seeberger, P. Org. React. 2006, 68, 303. doi:10.1002/0471264180.or064.02
- Vorm, Stefan van der; Hansen, Thomas; Hengst, Jacob M. A. van; S. Overkleeft, Herman; Marel, Gijsbert A. van der; C. Codée, Jeroen D. (2019). "Acceptor reactivity in glycosylation reactions". Chemical Society Reviews. 48 (17): 4688–4706. doi:10.1039/C8CS00369F. PMID 31287452.
- Vorm, S. van der; Hansen, T.; S. Overkleeft, H.; Marel, G. A. van der; C. Codée, J. D. (2017). "The influence of acceptor nucleophilicity on the glycosylation reaction mechanism". Chemical Science. 8 (3): 1867–1875. doi:10.1039/C6SC04638J. PMC 5424809. PMID 28553477.
- Wilhelm Koenigs and Edward Knorr (1901). "Ueber einige Derivate des Traubenzuckers und der Galactose (p )". Berichte der deutschen chemischen Gesellschaft. 34 (1): 957–981. doi:10.1002/cber.190103401162.
- Fischer, E. Ber. Dtsch. Chem. Ges. 1893, 26, 2400–2412
- Gervay, J., Hadd, M. J., J. Org. Chem. 1997, 62, 6961 – 6967.
- Hadd, M. J., Gervay, J. Carbohydr. Res. 1999, 320, 61 – 69.
- Miquel, N., Vignando, S., Russo, G., Lay, L. Synlett 2004, 2, 341 – 343.
- van Well, R. M., Kartha, K. P. R., Field, R. A. J. Carbohydr. Chem. 2005, 24, 463 – 474.
- Gervay, J., Nguyen, T. N., Hadd, M. J. Carbohydr. Res. 1997, 300, 119 – 125.
- Zhu, X. M., Schmidt, R. R. Angew. Chem. Int. Ed. 2009, 48, 1900 - 1934.
- Lam, S. N., Gervay-Hague, J. Org. Lett. 2003, 5, 4219 – 4222.
- Du, W., Gervay-Hague, J. Org. Lett. 2005, 7, 2063–2065.
- Du, W., Kulkarni, S. S., Gervay-Hague, J. Chem. Commun. 2007, 2336–2338.
- Fischer, E., Delbrück, K. Ber. Dtsch. Chem. Ges. 1909, 42, 1476–1482.
- Tai, C. A., Kulkarni, S. S., Hung, S. C. J. Org.Chem. 2003, 68, 8719 – 8722
- Agnihotri, G., Tiwari, P., Misra, A. K. Carbohydr. Res. 2005, 340, 1393–1396
- Hase-gawa, J. Y., Hamada, M., Miyamoto, T., Nishide, K., Kajimoto, T., Uenishi, J. I., Node, M. Carbohydr. Res. 2005, 340, 2360–2368.
- Veeneman, G. H., van Leeuwen, S. H., van Boom, J. H. Tetrahedron Lett. 1990, 31, 1331–1334.
- Schmidt, R. R., Michel, J. Angew. Chem. 1980, 92, 763 – 764.
- Schmidt, R. R., Michel, J. Angew. Chem. Int. Ed. Engl. 1980, 19, 731 – 732.
- Kale, R. R., McGannon, C. M., Fuller-Schaefer, C., Hatch, D. M., Flagler, M. J., Gamage, S. D., Weiss, A. A., Iyer, S. S. Angew. Chem. Int. Ed. 2008, 47, 1265 - 1268.
- Heuckendorff, Mads; Jensen, Henrik H. (2017-02-01). "Removal of some common glycosylation by-products during reaction work-up". Carbohydrate Research. 439: 50–56. doi:10.1016/j.carres.2016.12.007. ISSN 0008-6215.
- Joe, M., Bai, Y., Nacario, R. C., Lowary, T. L. J. Am. Chem. Soc. 2007, 129, 9885 - 9901.
- Wu, X. Y., Bundle, D. R. J. Org. Chem. 2005, 70, 7381 - 7388.
- Marco Brito-Arias, Síntesis and Characterization of Glycosides, second edition, Editorial Springer 2016.