Consensus CDS Project
The Consensus Coding Sequence (CCDS) Project is a collaborative effort to maintain a dataset of protein-coding regions that are identically annotated on the human and mouse reference genome assemblies. The CCDS project tracks identical protein annotations on the reference mouse and human genomes with a stable identifier (CCDS ID), and ensures that they are consistently represented by the National Center for Biotechnology Information (NCBI), Ensembl, and UCSC Genome Browser.[1] The integrity of the CCDS dataset is maintained through stringent quality assurance testing and on-going manual curation.[2]
| Content | |
|---|---|
| Description | Convergence towards a standard set of gene annotations |
| Contact | |
| Research center | National Center for Biotechnology Information European Bioinformatics Institute University of California, Santa Cruz Wellcome Trust Sanger Institute |
| Authors | Pruitt KD |
| Primary citation | Pruitt KD, et al (2009)[1] |
| Release date | 2009 |
| Access | |
| Website | https://www.ncbi.nlm.nih.gov/projects/CCDS/CcdsBrowse.cgi |
| Miscellaneous | |
| Version | CCDS Release 21 |
Motivation and background
Biological and biomedical research has come to rely on accurate and consistent annotation of genes and their products on genome assemblies. Reference annotations of genomes are available from various sources, each with their own independent goals and policies, which results in some annotation variation.
The CCDS project was established to identify a gold standard set of protein-coding gene annotations that are identically annotated on the human and mouse reference genome assemblies by the participating annotation groups. The CCDS gene sets that have been arrived at by consensus of the different partners [2] now consist of over 18,000 human and over 20,000 mouse genes (see CCDS release history). The CCDS dataset is increasingly representing more alternative splicing events with each new release.[3]
Contributing groups
Participating annotation groups include:[3]
- National Center for Biotechnology Information (NCBI)
- European Bioinformatics Institute (EBI)
- Wellcome Trust Sanger Institute (WTSI)
- HUGO Gene Nomenclature Committee (HGNC)
- Mouse Genome Informatics (MGI)
Manual annotation is provided by:
Defining the CCDS gene set
"Consensus" is defined as protein-coding regions that agree at the start codon, stop codon, and splice junctions, and for which the prediction meets quality assurance benchmarks.[1] A combination of manual and automated genome annotations provided by (NCBI) and Ensembl (which incorporates manual HAVANA annotations) are compared to identify annotations with matching genomic coordinates.
Quality assurance testing
In order to ensure that CDSs are of high quality, multiple quality assurance (QA) tests are performed (Table 1). All tests are performed following the annotation comparison step of each CCDS build and are independent of individual annotation group QA tests performed prior to the annotation comparison.[3]
| QA test | Purpose of the test |
|---|---|
| Subject to NMD | Checks for transcripts that may be subject to nonsense-mediated decay (NMD) |
| Low quality | Checks for low coding propensity |
| Non-consensus splice sites | Checks for non-canonical splice sites |
| Predicted pseudogene | Checks for genes that are predicted to be pseudogenes by UCSC |
| Too short | Checks for transcripts or proteins that are unusually short, typically <100 amino acids |
| Ortholog not found/not conserved | Checks for genes that are not conserved and/or are not in a HomoloGene cluster |
| CDS start or stop not in alignment | Checks for a start or stop codon in the reference genome sequence |
| Internal stop | Checks for the presence of an internal stop codon in the genomic sequence |
| NCBI:Ensembl protein length different | Checks if the protein encoded by the NCBI RefSeq is the same length as the EBI/WTSI protein |
| NCBI:Ensembl low percent identity | Checks for >99% overall identity between the NCBI and EBI/WTSI proteins |
| Gene discontinued | Checks if the GeneID is no longer valid |
Annotations that fail QA tests undergo a round of manual checking that may improve results or reach a decision to reject annotation matches based on QA failure.
Review process
The CCDS database is unique in that the review process must be carried out by multiple collaborators, and agreement must be reached before any changes can be made. This is made possible with a collaborator coordination system that includes a work process flow and forums for analysis and discussion. The CCDS database operates an internal website that serves multiple purposes including curator communication, collaborator voting, providing special reports and tracking the status of CCDS representations. When a collaborating CCDS group member identifies a CCDS ID that may need review, a voting process is employed to decide on the final outcome.
Manual curation
Coordinated manual curation is supported by a restricted-access website and a discussion e-mail list. CCDS curation guidelines were established to address specific conflicts that were observed at a higher frequency. Establishment of CCDS curation guidelines has helped to make the CCDS curation process more efficient by reducing the number of conflicting votes and time spent in discussion to reach a consensus agreement. A link to the CCDS curation guidelines can be found here.
Curation policies established for the CCDS data set have been integrated in to the RefSeq and HAVANA annotation guidelines and thus, new annotations provided by both groups are more likely to be concordant and result in addition of a CCDS ID. These standards address specific problem areas, are not a comprehensive set of annotation guidelines, and do not restrict the annotation policies of any collaborating group.[2] Examples include, standardized curation guidelines for selection of the initiation codon and interpretation of upstream ORFs and transcripts that are predicted to be candidates for nonsense-mediated decay. Curation occurs continuously, and any of the collaborating centers can flag a CCDS ID as a potential update or withdrawal.
Conflicting opinions are addressed by consulting with scientific experts or other annotation curation groups such as the HUGO Gene Nomenclature Committee (HGNC) and Mouse Genome Informatics (MGI). If a conflict cannot be resolved, then collaborators agree to withdraw the CCDS ID until more information becomes available.
Curation challenges and annotation guidelines
Nonsense-mediated decay (NMD): NMD is the most powerful mRNA surveillance process. NMD eliminates defective mRNA before it can be translated into protein.[4] This is important because if the defective mRNA is translated, the truncated protein may cause disease. Different mechanisms have been proposed to explain NMD; one being the exon junction complex (EJC) model. In this model, if the stop codon is >50 nt upstream of the last exon-exon junction, the transcript is assumed to be a NMD candidate.[2] The CCDS collaborators use a conservative method, based on the EJC model, to screen mRNA transcripts. Any transcripts determined to be NMD candidates are excluded from the CCDS data set except in the following situations:[2]
- all transcripts at one particular locus are assessed to be NMD candidates however the locus is previously known to be protein coding region;
- there is experimental evidence suggesting that a functional protein is produced from the NMD candidate transcript.
Previously, NMD candidate transcripts were considered to be protein coding transcripts by both RefSeq and HAVANA, and thereby, these NMD candidate transcripts were represented in the CCDS data set. The RefSeq group and the HAVANA project have subsequently revised their annotation policies.
Multiple in-frame translation start sites: Multiple factors contribute to translation initiation, such as upstream open reading frames (uORFs), secondary structure and the sequence context around the translation initiation site. A common start site is defined within Kozak consensus sequence: (GCC) GCCACCAUGG in vertebrates. The sequence in brackets (GCC) is the motif with unknown biological impact.[5] There are variations within Kozak consensus sequence, such as G or A is observed three nucleotides upstream (at position -3) of AUG. Bases between positions -3 and +4 of Kozak sequence have the most significant impact on translational efficiency. Hence, a sequence (A/G)NNAUGG is defined as a strong Kozak signal in the CCDS project.
According to the scanning mechanism, the small ribosomal subunit can initiate translation from the first reached start codon. There are exceptions to the scanning model:
- when the initiation site is not surrounded by a strong Kozak signal, which results in leaky scanning. Thereby, the ribosome skips this AUG and initiates translation from a downstream start site;
- when a shorter ORF can allow the ribosome to re-initiate translation at a downstream ORF.[5]
According to the CCDS annotation guidelines, the longest ORF must be annotated except when there is experimental evidence that an internal start site is used to initiate translation. Additionally, other types of new data, such as ribosome profiling data,[6] can be used to identify start codons. The CCDS data set records one translation initiation site per CCDS ID. Any alternative start sites may be used for translation and will be stated in a CCDS public note.
Upstream open reading frames: AUG initiation codons located within transcript leaders are known as upstream AUGs (uAUGs). Sometimes, uAUGs are associated with uORFs . uORFs are found in approximately 50% of human and mouse transcripts.[7] The existence of uORFs are another challenge for the CCDS data set. The scanning mechanism for translation initiation suggests that small ribosomal subunits (40S) bind at the 5’ end of a nascent mRNA transcript and scan for the first AUG start codon.[5] It is possible that an uAUG is recognised first, and the corresponding uORF is then translated. The translated uORF could be a NMD candidate, although studies have shown that some uORFs can avoid NMD. The average size limit for uORFs that will escape NMD is approximately 35 amino acids.[2][8] It also has been suggested that uORFs inhibit translation of the downstream gene by trapping a ribosome initiation complex and causing the ribosome to dissociate from the mRNA transcript before it reaches the protein-coding regions.[4][7] Currently, no studies have reported the global impact of uORFs on translational regulation.
The current CCDS annotation guidelines allow the inclusion of mRNA transcripts containing uORFs if they meet the following two biological requirements:[2]
- the mRNA transcript has a strong Kozak signal;
- the mRNA transcript is either ≥ 35 amino acids or overlaps with the primary open reading frame.
Read-through transcripts: Read-through transcripts are also known as conjoined genes or co-transcribed genes. Read-through transcripts are defined as transcripts combining at least part of one exon from each of two or more distinct known (partner) genes which lie on the same chromosome in the same orientation.[9] The biological function of read-through transcripts and their corresponding protein molecules remain unknown. However, the definition of a read-through gene in the CCDS data set is that the individual partner genes must be distinct, and the read-through transcripts must share ≥ 1 exon (or ≥ 2 splice sites except in the case of a shared terminal exon) with each of the distinct shorter loci.[2] Transcripts are not considered to be read-through transcripts in the following circumstances:
- when transcripts are produced from overlapping genes but do not share same splice sites;
- when transcripts are translated from genes that have nested structures relative to each other. In this instance, the CCDS collaborators and the HGNC have agreed that the read-through transcript be represented as a separate locus.
Quality of reference genome sequence: As the CCDS data set is built to represent genomic annotations of human and mouse, the quality problems with the human and mouse reference genome sequences become another challenge. Quality problems occur when the reference genome is misassembled. Thereby the misassembled genome may contain premature stop codons, frame-shift indels, or likely polymorphic pseudogenes. Once these quality problems are identified, the CCDS collaborators report the issues to the Genome Reference Consortium, which investigates and makes the necessary corrections.
Access to CCDS data
The CCDS project is available from the NCBI CCDS data set page (here), which provides FTP download links and a query interface to acquire information about CCDS sequences and locations. CCDS reports can be obtained by using the query interface, which is located at the top of the CCDS data set page. Users can select various types of identifiers such as CCDS ID, gene ID, gene symbol, nucleotide ID and protein ID to search for specific CCDS information.[1] The CCDS reports (Figure 1) are presented in a table format, providing links to specific resources, such as a history report, Entrez Gene [10] or re-query the CCDS data set. The sequence identifiers table presents transcript information in VEGA, Ensembl and Blink. The chromosome location table includes the genomic coordinates for each individual exon of the specific coding sequence. This table also provides links to several different genome browsers, which allow you to visualise the structure of the coding region.[1] Exact nucleotide sequence and protein sequence of the specific coding sequence are also displayed in the section of CCDS sequence data.
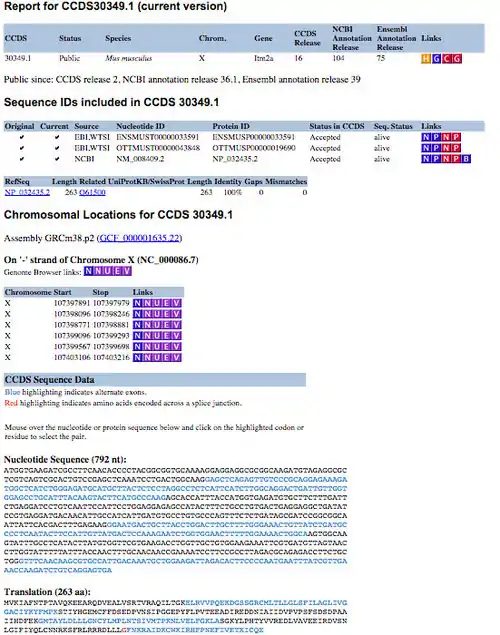
Current applications
The CCDS dataset is an integral part of the GENCODE gene annotation project[11] and it is used as a standard for high-quality coding exon definition in various research fields, including clinical studies, large-scale epigenomic studies, exome projects and exon array design.[3] Due to the consensus annotation of CCDS exons by the independent annotation groups, exome projects in particular have regarded CCDS coding exons as reliable targets for downstream studies (e.g., for single nucleotide variant detection), and these exons have been used as coding region targets in commercially available exome kits.[12]
CCDS release history
The CCDS data set size has continued to increase with both the computational genome annotation updates, which integrate new data sets submitted to the International Nucleotide Sequence Database Collaboration (INSDC), and on ongoing curation activities that supplement or improve upon that annotation. Table 2 summarises the key statistics for each CCDS build where Public CCDS IDs are all those that were not under review or pending an update or withdrawal at the time of the current release date.
| Release | Species | Assembly name | Public CCDS ID count | Gene ID count | Current release date |
|---|---|---|---|---|---|
| 1 | Homo sapiens | NCBI35 | 13,740 | 12,950 | Mar 14, 2007 |
| 2 | Mus musculus | MGSCv36 | 13,218 | 13,012 | Nov 28, 2007 |
| 3 | Homo sapiens | NCBI36 | 17,494 | 15,805 | May 1, 2008 |
| 4 | Mus musculus | MGSCv37 | 17, 082 | 16,888 | Jan 24, 2011 |
| 5 | Homo sapiens | NCBI36 | 19,393 | 17,053 | Sep 2, 2009 |
| 6 | Homo sapiens | GRCh37 | 22,912 | 18,174 | Apr 20, 2011 |
| 7 | Mus musculus | MGSCv37 | 21,874 | 19,507 | Aug 14, 2012 |
| 8 | Homo sapiens | GRCh37.p2 | 25,354 | 18,407 | Sep 6, 2011 |
| 9 | Homo sapiens | GRCh37.p5 | 26,254 | 18,474 | Oct 25, 2012 |
| 10 | Mus musculus | GRCm38 | 22,934 | 19,945 | Aug 5, 2013 |
| 11 | Homo sapiens | GRCh37.p9 | 27,377 | 18,535 | Apr 29, 2013 |
| 12 | Homo sapiens | GRCh37.p10 | 27,655 | 18,607 | Oct 24, 2013 |
| 13 | Mus musculus | GRCm38.p1 | 23,010 | 19,990 | Apr 7, 2014 |
| 14 | Homo sapiens | GRCh37.p13 | 28,649 | 18,673 | Nov 29, 2013 |
| 15 | Homo sapiens | GRCh37.p13 | 28,897 | 18,681 | Aug 7, 2014 |
| 16 | Mus musculus | GRCm38.p2 | 23,835 | 20,079 | Sep 10, 2014 |
| 17 | Homo sapiens | GRCh38 | 30,461 | 18,800 | Sep 10, 2014 |
| 18 | Homo sapiens | GRCh38.p2 | 31,371 | 18,826 | May 12, 2015 |
| 19 | Mus musculus | GRCm38.p3 | 24,834 | 20,215 | July 30, 2015 |
| 20 | Homo sapiens | GRCh38.p7 | 32,524 | 18,892 | Sep 8, 2016 |
| 21 | Mus musculus | GRCm38.p4 | 25,757 | 20,354 | Dec 8, 2016 |
The complete set of release statistics can be found at the official CCDS website on their Releases & Statistics page.
Future prospects
Long-term goals include the addition of attributes that indicate where transcript annotation is also identical (including the UTRs) and to indicate splice variants with different UTRs that have the same CCDS ID. It is also anticipated that as more complete and high-quality genome sequence data become available for other organisms, annotations from these organisms may be in scope for CCDS representation.
The CCDS set will become more complete as the independent curation groups agree on cases where they initially differ, as additional experimental validation of weakly supported genes occurs, and as automatic annotation methods continue to improve. Communication among the CCDS collaborating groups is ongoing and will resolve differences and identify refinements between CCDS update cycles. Human updates are expected to occur roughly every 6 months and mouse releases yearly.[3]
See also
- GENCODE
- Human Genome
- Mouse Genome Informatics
- RefSeq
- Ensembl
References
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, Hart E, Suner MM, Landrum MJ, Aken B, Ayling S, Baertsch R, Fernandez-Banet J, Cherry JL, Curwen V, Dicuccio M, Kellis M, Lee J, Lin MF, Schuster M, Shkeda A, Amid C, Brown G, Dukhanina O, Frankish A, Hart J, Maidak BL, Mudge J, Murphy MR, Murphy T, Rajan J, Rajput B, Riddick LD, Snow C, Steward C, Webb D, Weber JA, Wilming L, Wu W, Birney E, Haussler D, Hubbard T, Ostell J, Durbin R, Lipman D (2009). "The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes". Genome Res. 19 (7): 1316–23. doi:10.1101/gr.080531.108. PMC 2704439. PMID 19498102.
- Harte, RA; Farrell, CM; Loveland, JE; Suner, MM; Wilming, L; Aken, B; Barrell, D; Frankish, A; Wallin, C; Searle, S; Diekhans, M; Harrow, J; Pruitt, KD (2012). "Tracking and coordinating an international curation effort for the CCDS project". Database. 2012: bas008. doi:10.1093/database/bas008. PMC 3308164. PMID 22434842.
- Farrell, CM; O'Leary, NA; Harte, RA; Loveland, JE; Wilming, LG; Wallin, C; Diehans, M; Barrell, D; Searle, SM; Aken, B; Hiatt, SM; Frankish, A; Suner, MM; Rajput, B; Steward, CA; Brown, GR; Bennet, R; Murphy, M; Wu, W; Kay, MP; Hart, J; Rajan, J; Weber, J; Snow, C; Riddick, LD; Hunt, T; Webb, D; Thomas, M; Tamez, P; Rangwala, SH; McGarvey, KM; Pujar, S; Shkeda, A; Mudge, JM; Gonzale, JM; Gilbert, JG; Trevaion, SJ; Baetsch, R; Harrow, JL; Hubbard, T; Ostell, JM; Haussler, D; Pruitt, KD (2014). "Current status and new features of the Consensus Coding Sequence database". Nucleic Acids Res. 42 (D1): D865–D872. doi:10.1093/nar/gkt1059. PMC 3965069. PMID 24217909.
- Alberts, B; Johnson, A; Lewis, J; Raff, M; Roberts, K; Walter, P (2002). Molecular Biology of the Cell 5th edn. New York: Garland Science.
- Kozak, M (2002). "Pushing the limits of the scanning mechanism for initiation of translation". Gene. 299 (1–2): 1–34. doi:10.1016/S0378-1119(02)01056-9. PMC 7126118. PMID 12459250.
- Ingolia, NT; Brar, GA; Rouskin, S; McGeachy, AM; Weissman, JS (2014). "Genome-wide Annotation and Quantitation of Translation by Ribosome Profiling". Curr. Protoc. Mol. Biol. Chapter 4: Unit–4.18. doi:10.1002/0471142727.mb0418s103. ISBN 9780471142720. PMC 3775365. PMID 23821443.
- Calvo, SE; Pagliarni, DJ; Mootha, VK (2009). "Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans" (PDF). Proc. Natl. Acad. Sci. U.S.A. 106 (18): 7507–12. Bibcode:2009PNAS..106.7507C. doi:10.1073/pnas.0810916106. PMC 2669787. PMID 19372376.
- Silva, AL; Pereira, FJC; Morgado, A; Kong, J; Martins, R; Faustino, P; Liebhaber, SA; Romao, L (2006). "The canonical UPF1-dependent nonsense-mediated mRNA decay is inhibited in transcripts carrying a short open reading frame independent of sequence context". RNA. 12 (12): 2160–70. doi:10.1261/rna.201406. PMC 1664719. PMID 17077274.
- Prakash, Tulika; Sharma, Vineet K.; Adati, Naoki; Ozawa, Ritsuko; Kumar, Naveen; Nishida, Yuichiro; Fujikake, Takayoshi; Takeda, Tadayuki; Taylor, Todd D.; Michalak, Pawel (12 October 2010). "Expression of Conjoined Genes: Another Mechanism for Gene Regulation in Eukaryotes". PLOS ONE. 5 (10): e13284. Bibcode:2010PLoSO...513284P. doi:10.1371/journal.pone.0013284. PMC 2953495. PMID 20967262.
- Maglott, D.; Ostell, J.; Pruitt, K. D.; Tatusova, T. (28 November 2010). "Entrez Gene: gene-centered information at NCBI". Nucleic Acids Res. 39 (Database): D52–D57. doi:10.1093/nar/gkq1237. PMC 3013746. PMID 21115458.
- Harrow, J.; Frankish, A.; Gonzalez, J. M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B. L.; Barrell, D.; Zadissa, A.; Searle, S.; Barnes, I.; Bignell, A.; Boychenko, V.; Hunt, T.; Kay, M.; Mukherjee, G.; Rajan, J.; Despacio-Reyes, G.; Saunders, G.; Steward, C.; Harte, R.; Lin, M.; Howald, C.; Tanzer, A.; Derrien, T.; Chrast, J.; Walters, N.; Balasubramanian, S.; Pei, B.; Tress, M.; Rodriguez, J. M.; Ezkurdia, I.; van Baren, J.; Brent, M.; Haussler, D.; Kellis, M.; Valencia, A.; Reymond, A.; Gerstein, M.; Guigo, R.; Hubbard, T. J. (5 September 2012). "GENCODE: The reference human genome annotation for The ENCODE Project". Genome Res. 22 (9): 1760–1774. doi:10.1101/gr.135350.111. PMC 3431492. PMID 22955987.
- Parla, Jennifer S; Iossifov, Ivan; Grabill, Ian; Spector, Mona S; Kramer, Melissa; McCombie, W Richard (2011). "A comparative analysis of exome capture". Genome Biol. 12 (9): R97. doi:10.1186/gb-2011-12-9-r97. PMC 3308060. PMID 21958622.