Flory–Fox equation
Flory function
Synonym: viscosity function, , SI unit: mol−1

Coefficient connecting the intrinsic viscosity, the mean-square radius of gyration, and the molar mass of a chain macromolecule, according to the equation
- <>[1]



Kirkwood–Riseman theory
Theory, based on the pearl-necklace model, that describes the translational diffusive and viscous flows of an isolated linear macromolecule in solution in the theta state and accounts for the gradual change from freedraining behaviour at lower molecular weights to impermeable behaviour at higher molecular weights.[1]
Note |
|---|
|
The Kirkwood–Riseman theory is usually applied in the impermeable limit when it essentially relates the equivalent hydrodynamic radius to the root-mean-square unperturbed radius of gyration, <>, with <>
and
<>
where and are the equivalent hydrodynamic radii in translational diffusive flow and viscous flow. |






Flory–Fox assumption
Assumption that the Kirkwood–Riseman theory can be applied to linear isolated macromolecules in solution, independent of whether they are in the theta state.[1]
Note |
|---|
|
The Kirkwood–Riseman relationships between and and the root-mean-square radius of gyration, <>, are then <><>
and
<><>
where and are the equivalent hydrodynamic radii in translational diffusive flow and viscous flow, and and are the corresponding expansion factors. |






In polymer chemistry and polymer physics, the Flory–Fox equation is a simple empirical formula that relates molecular weight to the glass transition temperature of a polymer system. The equation was first proposed in 1950 by Paul J. Flory and Thomas G. Fox while at Cornell University.[2] Their work on the subject overturned the previously held theory that the glass transition temperature was the temperature at which viscosity reached a maximum. Instead, they demonstrated that the glass transition temperature is the temperature at which the free space available for molecular motions achieved a minimum value.[3] While its accuracy is usually limited to samples of narrow range molecular weight distributions, it serves as a good starting point for more complex structure-property relationships.
Overview
The Flory–Fox equation relates the number-average molecular weight, Mn, to the glass transition temperature, Tg, as shown below:

where Tg,∞ is the maximum glass transition temperature that can be achieved at a theoretical infinite molecular weight and K is an empirical parameter that is related to the free volume present in the polymer sample. It is this concept of “free volume” that is observed by the Flory–Fox equation.
Free volume can be most easily understood as a polymer chain's “elbow room” in relation to the other polymer chains surrounding it. The more elbow room a chain has, the easier it is for the chain to move and achieve different physical conformations. Free volume decreases upon cooling from the rubbery state until the glass transition temperature at which point it reaches some critical minimum value and molecular rearrangement is effectively “frozen” out, so the polymer chains lack sufficient free volume to achieve different physical conformations. This ability to achieve different physical conformations is called segmental mobility.
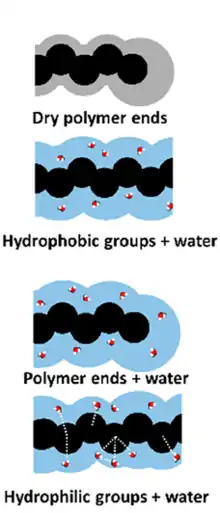
Free volume not only depends on temperature, but also on the number of polymer chain ends present in the system. End chain units exhibit greater free volume than units within the chain because the covalent bonds that make up the polymer are shorter than the intermolecular nearest neighbor distances found at the end of the chain. In other words, chain end units are less dense than the covalently bonded interchain units. This means that a polymer sample with long chain lengths (high molecular weights) will have fewer chain ends per total units and less free volume than a polymer sample consisting of short chains. In short, chain ends can be viewed as an “impurity” when considering the packing of chains, and more impurity results in a lower Tg. Recent computer simulation study showed that the classical picture of mobility around polymer chain can differ in the presence of plasticizer, especially if molecules of plasticizer can create hydrogen bonds with specific sites of the polymer chain, such as hydrophilic or hydrophobic groups. In such the case, polymer chain ends exhibit only a mere increase of the associated free volume as compared to the average associated free volume around main chain monomers. In special cases, the free volume around hydrophilic main chain sites can exceed the free volume associated to the hydrophilic polymer ends.[4]
Thus, glass transition temperature is dependent on free volume, which in turn is dependent on the average molecular weight of the polymer sample. This relationship is described by the Flory–Fox equation. Low molecular weight values result in lower glass transition temperatures whereas increasing values of molecular weight result in an asymptotic approach of the glass transition temperature to Tg,∞ . The figure to the left clearly displays this relationship – as molecular weight increases, the glass transition temperature increases asymptotically to Tg,∞ (in this arbitrary case shown in the image, Tg,∞ = 365 K).
Molecular-level derivation
The main shortcoming related to the free volume concept is that it is not so well defined at the molecular level. A more precise, molecular-level derivation of the Flory–Fox equation has been developed by Alessio Zaccone and Eugene Terentjev.[5] The derivation is based on a molecular-level model of the temperature-dependent shear modulus G of glassy polymers. The shear modulus of glasses has two main contributions,[6] one which is related to affine displacements of the monomers in response to the macroscopic strain, which is proportional to the local bonding environment and also to the non-covalent van der Waals-type interactions, and a negative contribution that corresponds to random (nonaffine) monomer-level displacements due to the local disorder. Due to thermal expansion, the first (affine) term decreases abruptly near the glass transition temperature Tg because of the weakening of the non-covalent interactions, while the negative nonaffine term is less affected by temperature. Experimentally, it is observed indeed that G drops sharply by many orders of magnitude at or near Tg (it does not really drop to zero but to the much lower value of the rubber elasticity plateau). By setting at the point where the G drops abruptly and solving for Tg, one obtains the following relation:[5]


In this equation, is the maximum volume fraction, or packing fraction, occupied by the monomers at the glass transition if there were no covalent bonds, i.e. in the limit of average number of covalent bonds per monomer . If the monomers can be approximated as soft spheres, then as in the jamming of soft frictionless spheres.[7] In the presence of covalent bonds between monomers, as is the case in the polymer, the packing fraction is lowered, hence , where is a parameter that expresses the effect of topological constraints due to covalent bonds on the total packing fraction occupied by the monomers in a given polymer. Finally, the packing fraction occupied by the monomers in the absence of covalent bonds is related to via thermal expansion, according to , which comes from integrating the thermodynamic relation between thermal expansion coefficient and volume V, , where is the coefficient of thermal expansion of the polymer in the glassy state. Note the relation between packing fraction and total volume given by , where is the total number of monomers, with molecular volume , contained in the total volume of the material, which has been used above. Hence is the integration constant in , and it was found that for the case of polystyrene. Also, is the molecular weight of one monomer in the polymer chain.

















Hence the above equation clearly recovers the Flory–Fox equation with its dependence on the number average molecular weight , and provides a molecular-level meaning to the empirical parameters present in the Fox-Flory equation. Furthermore, it predicts that , i.e. that the glass transition temperature is inversely proportional to the thermal expansion coefficient in the glass state.


Alternative equations
While the Flory–Fox equation describes many polymers very well, it is more reliable for large values of Mn [8] and samples of narrow weight distribution. As a result, other equations have been proposed to provide better accuracy for certain polymers. For example:

This minor modification of the Flory–Fox equation, proposed by Ogawa,[9] replaces the inverse dependence on Mn with the square of the product of the number-average molecular weight, Mn , and weight-average molecular weight, Mw . Additionally, the equation:

was proposed by Fox and Loshaek,[10] and has been applied to polystyrene, polymethylmethacrylate, and polyisobutylene, among others.
However, it is important to note that despite the dependence of Tg on molecular weight that the Flory-Fox and related equations describe, molecular weight is not necessarily a practical design parameter for controlling Tg because the range over which it can be changed without altering the physical properties of the polymer due to molecular weight change is small.[8]
The Fox Equation
The Flory–Fox equation serves the purpose of providing a model for how glass transition temperature changes over a given molecular weight range. Another method to modify the glass transition temperature is to add a small amount of low molecular weight diluent, commonly known as a plasticizer, to the polymer. The presence of a low molecular weight additive increases the free volume of the system and subsequently lowers Tg , thus allowing for rubbery properties at lower temperatures. This effect is described by the Fox equation:

Where w1 and w2 are weight fractions of components 1 and 2, respectively. In general, the accuracy of the Fox equation is very good and it is commonly also applied to predict the glass transition temperature in (miscible) polymer blends and statistical copolymers.[8]
References
- Stepto, Robert; Chang, Taihyun; Kratochvíl, Pavel; Hess, Michael; Horie, Kazuyuki; Sato, Takahiro; Vohlídal, Jiří (2015). "Definitions of terms relating to individual macromolecules, macromolecular assemblies, polymer solutions, and amorphous bulk polymers (IUPAC Recommendations 2014)". Pure and Applied Chemistry. 87 (1): 71–120. doi:10.1515/pac-2013-0201. S2CID 98061698.
- Fox, T.G.; Flory, P.J. (1950), "Second-order transition temperatures and related properties of polystyrene", Journal of Applied Physics, 21 (6): 581–591, doi:10.1063/1.1699711
- Markovitz, Hershel (May–June 1978). "Thomas G. Fox 1921–1977". Rheologica Acta. 17 (3): 207–209. doi:10.1007/BF01535056. S2CID 97830348.
- Capponi, S.; Alvarez, F.; Racko, D. (2020), "Free Volume in a PVME Polymer–Water Solution", Macromolecules, 53 (12): 4770–4782, doi:10.1021/acs.macromol.0c00472
- Zaccone, A.; Terentjev, E. (2013). "Disorder-Assisted Melting and the Glass Transition in Amorphous Solids". Physical Review Letters. 110 (17): 178002. arXiv:1212.2020. doi:10.1103/PhysRevLett.110.178002. PMID 23679782. S2CID 15600577.
- Zaccone, A.; Scossa-Romano, E. (2011). "Approximate analytical description of the nonaffine response of amorphous solids". Physical Review B. 83 (18): 184205. arXiv:1102.0162. doi:10.1103/PhysRevB.83.184205.
- O'Hern, C. S.; Silbert, L. E.; Liu, A. J.; Nagel, S. R. (2003). "Jamming at zero temperature and zero applied stress: The epitome of disorder". Physical Review E. 68 (1 Pt 1): 011306. doi:10.1103/PhysRevE.68.011306. PMID 12935136.
- Hiemenz, Paul; Timothy Lodge (2007). Polymer Chemistry. Boca Raton, Florida: CRC Press. ISBN 978-1-57444-779-8.
- Ogawa (1992), "Effects of molecular weight on mechanical properties of polypropylene", Journal of Applied Polymer Science, 1869 (10): 1869–1871, doi:10.1002/app.1992.070441022
- Fox, T.G.; Loshaek, S. (1955), "Influence of molecular weight and degree of crosslinking on the specific volume and glass temperature of polymers", Journal of Polymer Science, 371 (80): 371–390, doi:10.1002/pol.1955.120158006