Hoyeraal–Hreidarsson syndrome
Hoyeraal–Hreidasson syndrome[2]) is a very rare multisystem X-linked recessive disorder characterized by excessively short telomeres and is considered a severe form of dyskeratosis congenita.[2][3] Being an X-linked disorder, Hoyeraal–Hreidasson syndrome primarily affects males. Patients typically present in early childhood with cerebellar hypoplasia, immunodeficiency, progressive bone marrow failure, and intrauterine growth restriction.[2] The primary cause of death in Hoyeraal–Hreidasson syndrome is bone marrow failure, but mortality from cancer and pulmonary fibrosis is also significant.[4][5][6]
| Hoyeraal–Hreidarsson syndrome | |
|---|---|
| Other names | Progressive pancytopenia-immunodeficiency-cerebellar hypoplasia syndrome[1] |
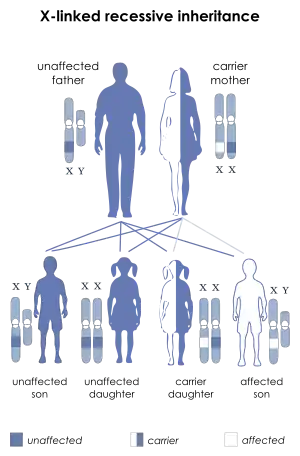 | |
| This condition is inherited in an X-linked recessive manner. | |
| Specialty | Medical genetics |
Presentation
The currently recognized features are cerebellar hypoplasia, immunodeficiency, progressive bone marrow failure, and intrauterine growth restriction. Patients also commonly exhibit symptoms such as microcephaly, aplastic anemia, and intellectual disability.[3]
Overlap with dyskeratosis congenita
Patients with Hoyeraal–Hreidasson syndrome frequently present with the mucocutaneous triad of nail dysplasia, lacy skin pigmentation, and oral leukoplakia.
Pathogenesis
Although the pathogenesis remains unknown, it is strongly suspected that the clinical sequelae of Hoyeraal–Hreidasson syndrome arise from the accelerated telomere shortening.[2] It has been associated with mutations in the poly(A)-specific ribonuclease poly(A)-specific ribonuclease gene.[7]
Diagnosis
- Neuroimaging – cerebellar hypoplasia/atrophy, small brainstem, thin corpus callosum and cerebral calcifications
- Molecular genetic testing – for confirmation
Treatment
Current treatment is supportive:
- The aplastic anemia and immunodeficiency can be treated by bone marrow transplantation.
- Supportive treatment for gastrointestinal complications and infections.
- Genetic counselling.
See also
References
- "Orphanet: Hoyeraal Hreidarsson syndrome". www.orpha.net. Retrieved 15 June 2019.
- Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA (August 2015). "Unraveling the pathogenesis of Hoyeraal–Hreidarsson syndrome, a complex telomere biology disorder". British Journal of Haematology. 170 (4): 457–71. doi:10.1111/bjh.13442. PMC 4526362. PMID 25940403.
- Knight SW, Heiss NS, Vulliamy TJ, Aalfs CM, McMahon C, Richmond P, et al. (November 1999). "Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal–Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1". British Journal of Haematology. 107 (2): 335–9. doi:10.1046/j.1365-2141.1999.01690.x. PMID 10583221. S2CID 23750791.
- Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, et al. (September 2013). "Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal–Hreidarsson syndrome". Proceedings of the National Academy of Sciences of the United States of America. 110 (36): E3408-16. Bibcode:2013PNAS..110E3408D. doi:10.1073/pnas.1300600110. PMC 3767560. PMID 23959892.
- Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, et al. (August 2013). "Human RTEL1 deficiency causes Hoyeraal–Hreidarsson syndrome with short telomeres and genome instability". Human Molecular Genetics. 22 (16): 3239–49. doi:10.1093/hmg/ddt178. PMID 23591994.
- Jullien L, Kannengiesser C, Kermasson L, Cormier-Daire V, Leblanc T, Soulier J, Londono-Vallejo A, de Villartay JP, Callebaut I, Revy P (May 2016). "Mutations of the RTEL1 Helicase in a Hoyeraal–Hreidarsson Syndrome Patient Highlight the Importance of the ARCH Domain". Human Mutation. 37 (5): 469–72. doi:10.1002/humu.22966. PMID 26847928. S2CID 21314739.
- Benyelles M, Episkopou H, O'Donohue MF, Kermasson L, Frange P, Poulain F, Burcu Belen F, Polat M, Bole-Feysot C, Langa-Vives F, Gleizes PE, de Villartay JP, Callebaut I, Decottignies A, Revy P (July 2019). "Impaired telomere integrity and rRNA biogenesis in PARN-deficient patients and knock-out models". EMBO Molecular Medicine. 11 (7): e10201. doi:10.15252/emmm.201810201. PMC 6609912. PMID 31273937.
External links
| Classification |
|---|