Illumina dye sequencing
Illumina dye sequencing is a technique used to determine the series of base pairs in DNA, also known as DNA sequencing. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris.[1][2] It was developed by Shankar Balasubramanian and David Klenerman of Cambridge University,[3] who subsequently founded Solexa, a company later acquired by Illumina. This sequencing method is based on reversible dye-terminators that enable the identification of single nucleotides as they are washed over DNA strands. It can also be used for whole-genome and region sequencing, transcriptome analysis, metagenomics, small RNA discovery, methylation profiling, and genome-wide protein-nucleic acid interaction analysis.[4][5]
Overview
Illumina sequencing technology works in three basic steps: amplify, sequence, and analyze. The process begins with purified DNA. The DNA is fragmented and adapters are added that contain segments that act as reference points during amplification, sequencing, and analysis. The modified DNA is loaded onto a flow cell where amplification and sequencing will take place. The flow cell contains nanowells that space out fragments and help with overcrowding.[6] Each nanowell contains oligonucleotides that provide an anchoring point for the adaptors to attach. Once the fragments have attached, a phase called cluster generation begins. This step makes about a thousand copies of each fragment of DNA and is done by bridge amplification PCR. Next, primers and modified nucleotides are washed onto the chip. These nucleotides have a reversible 3' fluorescent blocker so the DNA polymerase can only add one nucleotide at a time onto the DNA fragment.[6] After each round of synthesis, a camera takes a picture of the chip. A computer determines what base was added by the wavelength of the fluorescent tag and records it for every spot on the chip. After each round, non-incorporated molecules are washed away. A chemical deblocking step is then used to remove the 3’ fluorescent terminal blocking group.. The process continues until the full DNA molecule is sequenced.[5] With this technology, thousands of places throughout the genome are sequenced at once via massive parallel sequencing.
Procedure
Genomic Library
After the DNA is purified a DNA library, genomic library, needs to be generated. There are two ways a genomic library can be created, sonification and tagmentation. With tagmentation, transposases randomly cuts the DNA into sizes between 50 to 500 bp fragments and adds adaptors simultaneously.[6] A genetic library can also be generated by using sonification to fragment genomic DNA. Sonification fragments DNA into similar sizes using ultrasonic sound waves. Right and left adapters will need to be attached by T7 DNA Polymerase and T4 DNA ligase after sonification. Strands that fail to have adapters ligated are washed away.[7]
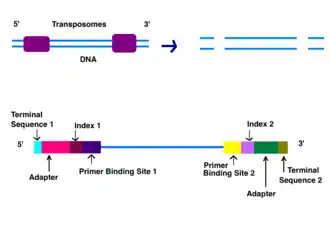
Adapters
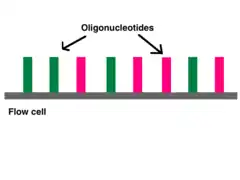
Adapters contain three different segments: the sequence complementary to solid support (oligonucleotides on flow cell), the barcode sequence (indices), and the binding site for the sequencing primer.[6] Indices are usually six base pairs long and are used during DNA sequence analysis to identify samples. Indices allow for up to 96 different samples to be run together, this is also known as multiplexing. During analysis, the computer will group all reads with the same index together.[8][9] Illumina uses a "sequence by synthesis" approach.[9] This process takes place inside of an acrylamide-coated glass flow cell.[10] The flow cell has oligonucleotides (short nucleotide sequences) coating the bottom of the cell, and they serve as the solid support to hold the DNA strands in place during sequencing. As the fragmented DNA is washed over the flow cell, the appropriate adapter attaches to the complementary solid support.
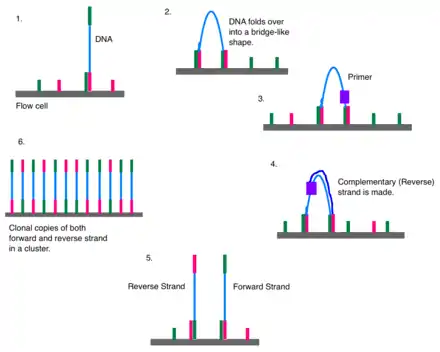
Bridge amplification
Once attached, cluster generation can begin. The goal is to create hundreds of identical strands of DNA. Some will be the forward strand; the rest, the reverse. This is why right and left adapters are used. Clusters are generated through bridge amplification. DNA polymerase moves along a strand of DNA, creating its complementary strand. The original strand is washed away, leaving only the reverse strand. At the top of the reverse strand there is an adapter sequence. The DNA strand bends and attaches to the oligo that is complementary to the top adapter sequence. Polymerases attach to the reverse strand, and its complementary strand (which is identical to the original) is made. The now double stranded DNA is denatured so that each strand can separately attach to an oligonucleotide sequence anchored to the flow cell. One will be the reverse strand; the other, the forward. This process is called bridge amplification, and it happens for thousands of clusters all over the flow cell at once.[11]
Clonal amplification
Over and over again, DNA strands will bend and attach to the solid support. DNA polymerase will synthesize a new strand to create a double stranded segment, and that will be denatured so that all of the DNA strands in one area are from a single source (clonal amplification). Clonal amplification is important for quality control purposes. If a strand is found to have an odd sequence, then scientists can check the reverse strand to make sure that it has the complement of the same oddity. The forward and reverse strands act as checks to guard against artefacts. Because Illumina sequencing uses DNA polymerase, base substitution errors have been observed,[12] especially at the 3' end.[13] Paired end reads combined with cluster generation can confirm an error took place. The reverse and forward strands should be complementary to each other, all reverse reads should match each other, and all forward reads should match each other. If a read is not similar enough to its counterparts (with which it should be a clone), an error may have occurred. A minimum threshold of 97% similarity has been used in some labs' analyses.[13]
Sequence by synthesis
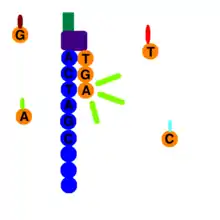
At the end of clonal amplification, all of the reverse strands are washed off the flow cell, leaving only forward strands. A primer attaches to the forward strands adapter primer binding site, and a polymerase adds a fluorescently tagged dNTP to the DNA strand. Only one base is able to be added per round due to the fluorophore acting as a blocking group; however, the blocking group is reversible.[6] Using the four-color chemistry, each of the four bases has a unique emission, and after each round, the machine records which base was added. Once the color is recorded the fluorophore is washed away and another dNTP is washed over the flow cell and the process is repeated. dATPs, dTTPs, dGTPs, and dCTPs are washed over the cell separately so each nucleotide is able to be identified.
Starting with the launch of the NextSeq and later the MiniSeq, Illumina introduced a new two-color sequencing chemistry. Nucleotides are distinguished by either one of two colors (red or green), no color ("black") or combining both colors (appearing orange as a mixture between red and green).
Once the DNA strand has been read, the strand that was just added is washed away. Then, the index 1 primer attaches, polymerizes the index 1 sequence, and is washed away. The strand forms a bridge again, and the 3' end of the DNA strand attaches to an oligo on the flow cell. The index 2 primer attaches, polymerizes the sequence, and is washed away.
A polymerase sequences the complementary strand on top of the arched strand. They separate, and the 3' end of each strand is blocked. The forward strand is washed away, and the process of sequence by synthesis repeats for the reverse strand.
Data analysis
The sequencing occurs for millions of clusters at once, and each cluster has ~1,000 identical copies of a DNA insert.[12] The sequence data is analyzed by finding fragments with overlapping areas, called contigs, and lining them up. If a reference sequence is known, the contigs are then compared to it for variant identification.
This piecemeal process allows scientists to see the complete sequence even though an unfragmented sequence was never run; however, because Illumina read lengths are not very long[13] (HiSeq sequencing can produce read lengths around 90 bp long[8]), it can be a struggle to resolve short tandem repeat areas.[8][12] Also, if the sequence is de novo and a reference doesn't exist, repeated areas can cause a lot of difficulty in sequence assembly.[12] Additional difficulties include base substitutions (especially at the 3' end of reads[13]) by inaccurate polymerases, chimeric sequences, and PCR-bias, all of which can contribute to generating an incorrect sequence.[13]
Comparison with other sequencing methods
This technique offers several advantages over traditional sequencing methods such as Sanger sequencing. Sanger sequencing requires two reactions, one for the forward primer and another for the reverse primer. Unlike Illumina, Sanger sequencing uses fluorescently labeled dideoxynucleoside triphosphates (ddNTPs) to determine the sequence of the DNA fragment. ddNTPs are missing the 3' OH group and terminates DNA synthesis permanently.[6] In each reaction tube, dNTPs and ddNTPs are added, along with DNA polymerase and primers. The ratio of ddNTPs to dNTPs matter since the template DNA needs to be completely synthesized, and an overabundance of ddNTPs will create multiple fragments of the same size and position of the DNA template. When the DNA polymerase adds a ddNTP the fragment is terminated and a new fragment is synthesized. Each fragment synthesized is one nucleotide longer than the last. Once the DNA template has been completely synthesized, the fragments are separated by capillary electrophoresis. At the bottom of the capillary tube a laser excites the fluorescently labeled ddNTPs and a camera captures the color emitted.
Due to the automated nature of Illumina dye sequencing it is possible to sequence multiple strands at once and gain actual sequencing data quickly. With Sanger sequencing, only one strand is able to be sequenced at a time and is relatively slow. Illumina only uses DNA polymerase as opposed to multiple, expensive enzymes required by other sequencing techniques (i.e. pyrosequencing).[14]
Examples of use
Illumina sequencing has been used to research transcriptomes of the sweet potato[15] and the gymnosperm genus Taxus.[16]
References
- CA 2158975, Canard B, Sarfati S, "Novel derivatives usable for the sequencing of nucleic acids", published 13 October 1994, assigned to Pasteur Institute
- Canard B, Sarfati RS (October 1994). "DNA polymerase fluorescent substrates with reversible 3'-tags". Gene. 148 (1): 1–6. doi:10.1016/0378-1119(94)90226-7. PMID 7523248.
- "History of Illumina Sequencing". Archived from the original on 12 October 2014.
- "Illumina - Sequencing and array-based solutions for genetic research". www.illumina.com.
- Meyer M, Kircher M (June 2010). "Illumina sequencing library preparation for highly multiplexed target capture and sequencing". Cold Spring Harbor Protocols. 2010 (6): pdb.prot5448. doi:10.1101/pdb.prot5448. PMID 20516186.
- Clark, David P. (2 November 2018). Molecular biology. Pazdernik, Nanette Jean,, McGehee, Michelle R. (Third ed.). London. ISBN 978-0-12-813289-0. OCLC 1062496183.
- "Illumina Sequencing Technology". Retrieved 24 September 2015.
- Feng YJ, Liu QF, Chen MY, Liang D, Zhang P (January 2016). "Parallel tagged amplicon sequencing of relatively long PCR products using the Illumina HiSeq platform and transcriptome assembly". Molecular Ecology Resources. 16 (1): 91–102. doi:10.1111/1755-0998.12429. PMID 25959587. S2CID 36882760.
- Illumina, Inc. "Multiplexed Sequencing with the Illumina Genome Analyzer System" (PDF). Retrieved 25 September 2015.
- Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, et al. (July 2012). "A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers". BMC Genomics. 13: 341. doi:10.1186/1471-2164-13-341. PMC 3431227. PMID 22827831.
- Clark, David P.; Pazdernik, Nanette J.; McGehee, Michelle R. (2019). Molecular Biology. Academic Cell. pp. 253–255. ISBN 9780128132883.
- Morozova O, Marra MA (November 2008). "Applications of next-generation sequencing technologies in functional genomics". Genomics. 92 (5): 255–64. doi:10.1016/j.ygeno.2008.07.001. PMID 18703132.
- Jeon YS, Park SC, Lim J, Chun J, Kim BS (January 2015). "Improved pipeline for reducing erroneous identification by 16S rRNA sequences using the Illumina MiSeq platform". Journal of Microbiology. 53 (1): 60–9. doi:10.1007/s12275-015-4601-y. PMID 25557481. S2CID 17210846.
- Pettersson E, Lundeberg J, Ahmadian A (February 2009). "Generations of sequencing technologies". Genomics. 93 (2): 105–11. doi:10.1016/j.ygeno.2008.10.003. PMID 18992322.
- Wang Z, Fang B, Chen J, Zhang X, Luo Z, Huang L, et al. (December 2010). "De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas)". BMC Genomics. 11: 726. doi:10.1186/1471-2164-11-726. PMC 3016421. PMID 21182800.
- Hao DC, Ge G, Xiao P, Zhang Y, Yang L (22 June 2011). "The first insight into the tissue specific taxus transcriptome via Illumina second generation sequencing". PLOS ONE. 6 (6): e21220. Bibcode:2011PLoSO...621220H. doi:10.1371/journal.pone.0021220. PMC 3120849. PMID 21731678.