JUNQ and IPOD
JUNQ and IPOD are types of cytosolic protein inclusion bodies in eukaryotes.
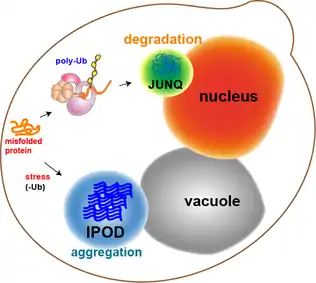
Neurodegenerative diseases, such as Parkinson's, Alzheimer's, and Huntington's, are associated and correlated with protein aggregation and accumulation of misfolded proteins in inclusion bodies. For many years, protein aggregation was considered a random process by which misfolded proteins stick to each other to form inclusions[1] (imagine a bundle of hairs haphazardly piling up in a corner of a room). Moreover, protein aggregates were thought to be toxic agents and the cause for neuronal dysfunction and death. However, recent studies, using advanced methods (i.e. fluorescence microscopy), show that protein aggregation may actually be a tightly regulated, organized process, by which the cell protects itself from toxic proteins by sequestration to inclusion bodies.[2] In 2008, Daniel Kaganovich showed that eukaryotic cells sort misfolded proteins into two distinct inclusion bodies in a well-managed cellular process:[3]
- The JUNQ (JUxta Nuclear Quality control compartment)
- The IPOD (Insoluble Protein Deposit)
JUNQ and IPOD are evolutionarily conserved, and are found in specific and defined cellular sites. Delivery of misfolded, aggregated proteins to JUNQ and IPOD require an intact cytoskeleton and specific cellular quality control components, such as Heat Shock Proteins (HSPs).[4] The partition into the two distinct inclusion bodies is due to the different handling and processing of different kinds of misfolded proteins (e.g. ubiquitinated vs. non-ubiquitinated proteins). Segregation of toxic protein aggregates into JUNQ and IPOD inclusion bodies is a means by which mammalian cells can be rejuvenated through asymmetric division.[5]
Thus, the discovery of JUNQ and IPOD provided a new striking perspective of how cells manage misfolded aggregated proteins and gave convincing proof that protein aggregation is a non-random, well regulated and controlled cellular process. Furthermore, the discovery of JUNQ and IPOD suggested that in addition to temporal quality control (i.e. time dependent administration of damaged proteins) cells exploit homeostasis spatially:[6] If degradation isn't available, protection of the cellular environment from a misfolded protein is accomplished by its sequestration to an aggregate inclusion.
Background
To function properly, most proteins must preserve a low-energy, three-dimensional structure known as the native state. The stability of a protein is tightly regulated through all its life stages: from cradle, as it is synthesized at the ribosome, through folding or assembly, till grave – when the protein is degraded and cleared from the cellular environment.[7] Protein homeostasis (proteostasis),[8] results from the coordinated action of the different arms of the cellular quality control system: molecular chaperones, proteases and other regulatory factors. Hence, cellular viability depends on timely and efficient management of misfolded proteins. Such management, by the quality control machinery, includes recognition of the misfolded protein by chaperones and E3 ligases, ubiquitination and degradation.
Proteostasis collapse, due to damage, stress, mutations, and aging, has been implicated as a basis for a large number of common human disorders, such as neurodegenerative diseases.[9] Although caused by different kinds of mutated proteins (e.g. in Huntington's disease – the protein Huntingtin) and disruptive to distinct tissues (e.g. in Huntington's disease – the striatum), such diseases share a common feature: accumulation of misfolded proteins in inclusion bodies. Thus, it was thought that the inclusion bodies are the cause of such diseases. However, the nature and characteristics of those intra-cellular inclusion bodies stayed elusive. Different kinds of proteins (e.g. prions, ERAD substrates) were reported to form different kinds of inclusion bodies (e.g. aggresomes, amyloids), yet it remained obscure if those observations combine into one and relate to the same sub-cellular site. Moreover, the pathways leading to inclusion formation and the involvement of the cellular protein quality control machinery were undefined and unknown. Thus, a systematic study providing a comprehensive understanding of protein aggregation and inclusion bodies was required. The discovery of JUNQ and IPOD[3] suggested new insights of how the cell manages different kinds of misfolded proteins and offered a novel framework for putting together the great puzzle of protein aggregation.[2]
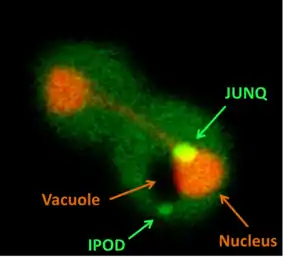
Discovery
The fate of misfolded proteins and the process leading to the formation of aggregate inclusions, were initially studied using biochemical methods (e.g. western blotting).
Deeper insights into the biological process of protein quality control and aggregation was made possible by a novel approach to looking at this problem, termed "Live Cell Imaging".[10]
Live cell imaging enables in vivo tracking of proteins in space and time, in their natural endogenous environment. Thus, such a method provides more information about the dynamics and stages of biological events and processes. The method takes advantage of the easily detectable fluorescent proteins fused to a protein of interest, which can then be followed inside a cell using a fluorescence microscope. The cell may then be treated by a perturbation of interest (e.g. a drug, expression of a misfolded protein), and various properties of the fluorescently tagged protein can be assayed using time-lapse microscopy:
- Changes of the fluorescence level indicates changes of expression levels (i.e. higher levels = upregulation of a protein)
- Changes of localization (e.g. entrance of a protein from the cytosol to the nucleus)
- Solubility (e.g. by using the FRAP assay[11])
- Interplay with the intracellular environment (e.g. by using the FLIP assay[11])
In order to monitor the fate of cytosolic misfolded proteins in vivo, a plasmid carrying a GFP tagged folding reporter was cloned. The folding reporter, a model protein for aggregation, was a Ubc9 (SUMO-conjugating enzyme) mutant (UBC9ts), harboring a missense mutation (Y68L) with a temperature- sensitive (ts) phenotype.[12][13] The marginally stable Ubc9ts is fully functional under physiological permissive conditions (25 °C) due to active cellular chaperones. The GFP–Ubc9ts was transformed into yeast and visualized using a fluorescence microscope.
Monitoring the folding sensor GFP–Ubc9ts was thought to indicate the cellular proteostasis, and to assay the ability of the cellular protein quality control system to deal with various kinds of stress. It was then observed that under normal conditions, GFP–Ubc9ts is diffused in the nucleus and in the cytosol. However, upon heat shock, GFP–Ubc9ts formed cytosolic punctate structures. Strikingly, when the proteasome was impaired and clearance of the misfolded protein by degradation was blocked, two distinct cytosolic inclusions were observed to be formed. Standard and conservative biochemical methods, such as cell fractionation and western blotting would not have revealed the partition into the two types of cytosolic aggregates.
The two detected inclusions were shown to be evolutionarily conserved quality control compartments, with different characteristics and distinct functions. They were named JUNQ (JUxta Nuclear Quality control compartment) and IPOD (Insoluble Protein Deposit),[3] and represent two cellular pathways for the sequestration and management of aggregation prone, potentially toxic proteins.
Partition of quality control substrates (i.e. misfolded proteins) to either compartment depends on their ubiquitination status and aggregation state (i.e. solubility):
Proteins that are ubiquitinated are delivered to the JUNQ, where they are processed for degradation by the proteasome. Misfolded proteins that are not ubiquitinated and terminally aggregated are sequestered to the IPOD.
Thus, the sub-cellular location of a misfolded protein (i.e. in the JUNQ or in the IPOD) provides information about its interaction with the cellular protein quality control machinery (e.g. its E3 ligase).
JUNQ
JUNQ is the JUxta Nuclear Quality control compartment.
_tethered_to_the_nucleus_(orange).tif.jpg.webp)
Significance
To maintain cellular homeostasis, the cellular quality control system must distinguish between folded and misfolded proteins. A misfolded protein will be recognized and tightly taken care of by either refolding or ubiquitination and proteasomal degradation.
However, cellular increase of misfolded protein loads, due to various kinds of stresses (e.g. heat shock), may saturate and exhaust the quality control machinery. In such cases, degradation of misfolded proteins is unavailable, and a second line of active cellular defense mechanism must be executed: directing misfolded proteins to specific cellular sites.[2]
The JUNQ serves as such a sequestration site. It was shown[3] that when the proteasome is impaired (e.g. by low expression levels of the proteasome subunit RPN11), ubiquitinated misfolded proteins are sorted into the JUNQ. Upon recovery from stress conditions (e.g. recovery from heat shock at a permissive temperature), misfolded proteins that accumulate in the JUNQ may be either refolded by the cellular chaperone machinery, or degraded by the 26S proteasome. Thus, the sequestration of a protein to the JUNQ is reversible.
Properties
The JUNQ is a non- membrane bound cellular site located in a margin of the nucleus, in close proximity to the endoplasmic reticulum. FRAP and FLIP assays revealed that proteins in the JUNQ are soluble and exchange with the cytosol, suggesting that the JUNQ has a dynamic structure.
Delivery to the JUNQ depends on molecular chaperones and co-chaperones and on the actin cytoskeleton.[4] Misfolded proteins must be ubiquitinated to be sorted to the JUNQ. If ubiquitination is blocked, a misfolded protein will be directed to the IPOD inclusion. Misfolded protein accumulation recruits 26S proteasomes to the JUNQ.
IPOD
IPOD is the Insoluble Protein Deposit compartment.
_tethered_to_the_vacuole_(green).tif.jpg.webp)
Significance
It is becoming more evident that the cellular capacity to maintain proteostasis[8] declines with age,[9] thereby causing the late onset of neurodegenerative diseases. In such diseases (e.g. Huntington's disease), a mutated protein misfolds and becomes toxic to the cellular environment by various ways such as denaturating cytosolic proteins.[14] Incompetent of degrading those toxic species, the cell must isolate them to avoid their hazardous interaction with the cellular proteome. The IPOD was shown[3] to be the sub-cellular site to which toxic amyloidogenic proteins are sequestered to, hereby serving as a protective quality control compartment.
In addition, it was suggested by the Lindquist group, that the IPOD is the site where yeast prions undergo a maturation process.[15] Thus, the IPOD may serve not only as a sequestration site, but also as a functional compartment.[15]
Properties
The IPOD is a non- membrane bound cellular site, which in yeast is located by the vacuole. FRAP and FLIP assays revealed that proteins in the IPOD are tightly packed, in-soluble and don't exchange with the cytosol. Amyloidogenic proteins, such as the Huntingtin protein, are the IPOD's substrates.
Misfolded proteins must be non-ubiquitinated to be sorted to the IPOD. Ubiquitination of an otherwise IPOD substrate, such as the RNQ1 fungal prion, will result in its sequestration in the JUNQ inclusion.
Upon accumulation of misfolded proteins, the disaggregase chaperone, AAA protein HSP104, localizes to the IPOD. It is yet to be determined if HSP104 functions in the IPOD or is simply sequestered there being hooked to a substrate.
The pre-autophagosomal structure (PAS) is localized by the IPOD.[16] However, it wasn't shown that IPOD substrates are delivered to the vacuole, and so the link between the IPOD and autophagy is yet to be determined.[3]
See also
References
- Treusch, Sebastian; Cyr, Douglas M.; Lindquist, Susan (2009). "Amyloid deposits: Protection against toxic protein species?" (PDF). Cell Cycle. 8 (11): 1668–74. doi:10.4161/cc.8.11.8503. PMC 4451085. PMID 19411847.
- Tyedmers, Jens; Mogk, Axel; Bukau, Bernd (2010). "Cellular strategies for controlling protein aggregation". Nature Reviews Molecular Cell Biology. 11 (11): 777–88. doi:10.1038/nrm2993. PMID 20944667. S2CID 22449895.
- Kaganovich, Daniel; Kopito, Ron; Frydman, Judith (2008). "Misfolded proteins partition between two distinct quality control compartments". Nature. 454 (7208): 1088–95. Bibcode:2008Natur.454.1088K. doi:10.1038/nature07195. PMC 2746971. PMID 18756251.
- Specht, S.; Miller, S. B. M.; Mogk, A.; Bukau, B. (2011). "Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae". The Journal of Cell Biology. 195 (4): 617–29. doi:10.1083/jcb.201106037. PMC 3257523. PMID 22065637.
- Ogrodnik M, Salmonowicz H, Brown R, Turkowska J, Sredniawa W, Pattabiraman S, Amen T, Abraham AC, Eichler N, Lyakhovetsky R, Kaganovich D (2014). "Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin". Proceedings of the National Academy of Sciences of the United States of America. 111 (22): 8049–54. Bibcode:2014PNAS..111.8049O. doi:10.1073/pnas.1324035111. PMC 4050583. PMID 24843142.
- Nystrom, T. (2010). "Spatial protein quality control and the evolution of lineage-specific ageing". Philosophical Transactions of the Royal Society B: Biological Sciences. 366 (1561): 71–5. doi:10.1098/rstb.2010.0282. PMC 3001311. PMID 21115532.
- Morimoto, R. I.; Cuervo, A. M. (2009). "Protein Homeostasis and Aging: Taking Care of Proteins from the Cradle to the Grave". The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 64A (2): 167–70. doi:10.1093/gerona/gln071. PMC 2655025. PMID 19228787.
- Powers, Evan T.; Morimoto, Richard I.; Dillin, Andrew; Kelly, Jeffery W.; Balch, William E. (2009). "Biological and Chemical Approaches to Diseases of Proteostasis Deficiency". Annual Review of Biochemistry. 78: 959–91. doi:10.1146/annurev.biochem.052308.114844. PMID 19298183.
- Ben-Zvi, A.; Miller, E. A.; Morimoto, R. I. (2009). "Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging". Proceedings of the National Academy of Sciences. 106 (35): 14914–9. Bibcode:2009PNAS..10614914B. doi:10.1073/pnas.0902882106. JSTOR 40484529. PMC 2736453. PMID 19706382.
- "Live-Cell Imaging".
- Lippincott-Schwartz, J.; Patterson, GH (2003). "Development and Use of Fluorescent Protein Markers in Living Cells". Science. 300 (5616): 87–91. Bibcode:2003Sci...300...87L. doi:10.1126/science.1082520. PMID 12677058. S2CID 7831723.
- Seufert, W.; Seufert, W (1996). "A Yeast Ubc9 Mutant Protein with Temperature-sensitive in Vivo Function is Subject to Conditional Proteolysis by a Ubiquitin- and Proteasome-dependent Pathway". Journal of Biological Chemistry. 271 (42): 25790–6. doi:10.1074/jbc.271.42.25790. PMID 8824207.
- Tongaonkar, Prasad; Beck, Konrad; Shinde, Ujwal P.; Madura, Kiran (1999). "Characterization of a Temperature-Sensitive Mutant of a Ubiquitin-Conjugating Enzyme and Its Use as a Heat-Inducible Degradation Signal". Analytical Biochemistry. 272 (2): 263–9. doi:10.1006/abio.1999.4190. PMID 10415098.
- England, Jeremy L.; Kaganovich, Daniel (2011). "Polyglutamine shows a urea-like affinity for unfolded cytosolic protein". FEBS Letters. 585 (2): 381–4. doi:10.1016/j.febslet.2010.12.023. PMID 21176779. S2CID 348468.
- Tyedmers, J.; Treusch, S.; Dong, J.; McCaffery, J. M.; Bevis, B.; Lindquist, S. (2010). "Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation". Proceedings of the National Academy of Sciences. 107 (19): 8633–8. Bibcode:2010PNAS..107.8633T. doi:10.1073/pnas.1003895107. PMC 2889312. PMID 20421488.
- Suzuki, K.; Kirisako, T; Kamada, Y; Mizushima, N; Noda, T; Ohsumi, Y (2001). "The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation". The EMBO Journal. 20 (21): 5971–81. doi:10.1093/emboj/20.21.5971. PMC 125692. PMID 11689437.