Non-linear effects
In enantioselective synthesis, a non-linear effect refers to a process in which the enantiopurity of the catalyst or chiral auxiliary does not correspond with the enantiopurity of the product produced. For example: a racemic catalyst would be expected to convert a prochiral substrate into a racemic product (a linear effect), but this is not always the case and a chirally enriched product can be produced instead (a non-linear effect).[1][2]
This can be expressed mathematically, as shown in Equation 1. Stereoselection that is higher or lower than the enantiomeric excess of the catalyst is considered non-ideal behavior. In non-ideal behavior, this deviation from linearity is described as the non-linear effect, NLE.[3]

For an ideal asymmetric reaction, the eeproduct may be described as the product of eemax multiplied by the eecatalyst. This is not the case for reactions exhibiting NLE's.[4]
Non-linear effects often arise in reactions with a scalemic catalyst composition.[3] As first observed by Wynberg and Feringa in 1976, different enantiomers of the chiral catalysts form heterochiral complexes, more specifically high order aggregates or dimeric forms of the catalyst.[5] These heterochiral complexes influence the effective stereoinduction of a scalemic catalyst. Additional sources of non-linear effects include autocatalysis, the process in which the reaction catalyzes itself.[6] General definitions and mathematical models are key to understanding non-linear effects and their application to specific chemical reactions. In the past two decades, the study of non-linear effects has shown to elucidate reaction mechanism and guide in synthetic applications.
Types of non-linear effects
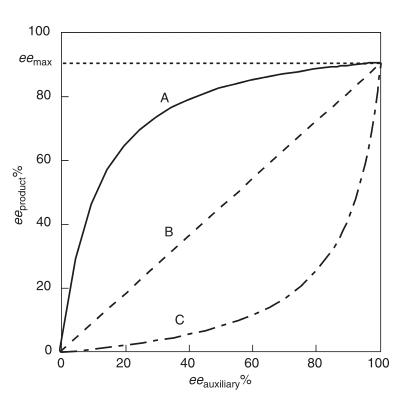
Positive non-linear effect, (+)-NLE
A positive non-linear effect, (+)-NLE, is present in an asymmetric reaction which demonstrates a higher product ee (eeproduct ) than predicted by an ideal linear situation (Figure 1).[4] It is often referred to as asymmetric amplification, a term coined by Oguni and co-workers.[4] An example of a positive non-linear effect is observed in the case of Sharpless epoxidation with the substrate geraniol.[7] In all cases of chemical reactivity exhibiting (+)-NLE, there is an innate tradeoff between overall reaction rate and enantioselectivity. The overall rate is slower and the enantioselectivity is higher relative to a linear behaving reaction.
Negative non-linear effect, (−)-NLE
Referred to as asymmetric depletion, a negative non-linear effect is present when the eeproduct is lower than predicted by an ideal linear situation.[3] In contrast to a (+)-NLE, a (−)-NLE results in a faster overall reaction rate and a decrease in enantioselectivity. Synthetically, a (−)-NLE effect could be beneficial with a reasonable assay for separating product enantiomers and a high output is necessary . An interesting example of a (−)-NLE effect has been reported in asymmetric sulfide oxidations.[8]
Modeling non-linear effects
1n 1986, Henri B. Kagan and coworkers observed a series of known reactions that followed a non-ideal behavior. A correction factor, f, was adapted to Equation 1 to fit the kinetic behavior of reactions with NLEs (Equation 2).[3]

Equation 2: A general mathematical equation that describes non-linear behavior[9]
Unfortunately, Equation 2 is too general to apply to specific chemical reactions. Due to this, Kagan and coworkers also developed simplified mathematical models to describe the behavior of catalysts which lead to non-linear effects.[3] These models involve generic MLn species, based on a metal (M) bound to n number of enantiomeric ligands (L). The type of MLn model varies among asymmetric reactions, based on the goodness of fit with reaction data. With accurate modeling, NLE may elucidate mechanistic details of an enantioselective, catalytic reaction.[8]
General description
The simplest model to describe a non-linear effect, the ML2 model involves a metal system (M) with two chiral ligands, LR and LS. In addition to the catalyzed reaction of interest, the model accounts for a steady state equilibrium between the unbound and bound catalyst complexes.[3] There are three possible catalytic complexes at equilibrium (MLSLR, MLSLS, MLRLR). The two enantiomerically pure complexes ( MLSLS, MLRLR) are referred to as homochiral complexes.[3] The possible heterochiral complex, MLRLS, is often referred to as a meso-complex.[3]

The equilibrium constant that describes this equilibrium, K, is presumably independent on the catalytic chemical reaction. In Kagan's model, K is determined by the amount of aggregation present in the chemical environment. A K=4 is considered to be the state at which there is a statistical distribution of ligands to each metal complex.[3] In other words, there is no thermodynamic disadvantage or advantage to the formation of heterochiral complexes at K=4.[4]
Obeying the same kinetic rate law, each of the three catalytic complexes catalyze the desired reaction to form product.[8] As enantiomers of each other, the homochiral complexes catalyze the reaction at the same rate, although opposite absolute configuration of the product is induced (i.e. rRR=rSS). The heterochiral complex, however, forms a racemic product at a different rate constant (i.e. rRS).[9]
Mathematical model for the ML2 Model
In order to describe the ML2 model in quantitative parameters, Kagan and coworkers described the following formula:

In the correction factor, Kagan and co-workers introduced two new parameters absent in Equation 1, β and g.[9] In general, these parameters represent the concentration and activity of three catalytic complexes relative to each other. β represents the relative amount of the heterochiral complex (MLRLS) as shown in Equation 3.[3] It is important to recognize that the equilibrium constant K is independent on both β and g.[8] As described by Donna Blackmond at Scripps Research Institute, "the parameter K is an inherent property of the catalyst mixture, independent of the eecatalyst. K is also independent of the catalytic reaction itself, and therefore independent of the parameter g."

Equation 3: The correction factor, β, may be described as z, the heterochiral complex concentration, divided by x and y, the respective concentrations of the complex concentration divided by x and y, the respective concentrations of the homochiral complexes [3]
The parameter g represents the reactivity of the heterochiral complex relative to the homochiral complexes. As shown in Equation 5, this may be described in terms of rate constants. Since the homochiral complexes react at identical rates, g can then be described as the rate constant corresponding to the heterochiral complex divided by the rate constant corresponding to either homochiral complex.

Equation 4: The correction parameter, g, can be described as the rate of product formation with the heterochiral catalyst MLRLS divided by the rate of product formation of the homochiral complex (MLRLR or MLSLS).
Interpretation of the mathematical results of the ML2 Model
- If β=0 or g=1, the ML2 equation simplifies to the Equation 1. No meso catalyst complex is present or active. Therefore, the simple additive properties should apply to such a scenario to establish a linear relationship between product enantioselectivity and the enantiopurity of the chiral catalyst.
- If the correction factor is greater than one, the reaction displays an asymmetric amplification, also known as a positive non-linear effect. Under the ML2 model, a (+)-NLE infers a less reactive heterochiral catalyst. In this case, the equilibrium constant K also increases as the correction factor increases. Although the product enantioselectivity is relatively high compared to the enantiopurity of the chiral catalyst, this comes at a cost of the overall reaction rate. In order to achieve an asymmetric amplification, there must be a relatively large concentration of the heterochiral complex. In addition, this heterochiral complex must have a substantially slower rate of reactivity, rRS. Therefore, the reactive catalytic species should decrease in concentration, leading to an overall slower reaction rate.
- If the correction factor is less than one, the reaction displays an asymmetric depletion, also known as a negative non-linear effect. In this scenario, the heterochiral catalyst is relatively more reactive than the homochiral catalyst complexes. In this case, the (−)-NLE may result in an overall faster although less selective product formation.
iv. Reaction Kinetics with the ML2 Model: Following H.B. Kagan's publication of the ML2 model, Professor Donna Blackmond at Scripps demonstrated how this model could be used to also calculate the overall reaction rates. With these relative reaction rates, Blackmond showed how the ML2 model could be used to formulate kinetic predictions which could then be compared to experimental data. The overall rate equation, Equation 6, is shown below.[8]

In addition to the goodness of fit to the model, kinetic information about the overall reaction may further validate the proposed reaction mechanism. For instance, a positive NLE in the ML2 should result in an overall lower reaction rate.[8] By solving the reaction rate from Equation 6, one can confirm if that is the case.
General description
Similar to the ML2 model, this modified system involves chiral ligands binding to a metal center (M) to create a new center of chirality.[4] There are four pairs of enantiomeric chiral complexes in the M*L2 model, as shown in Figure 5.

In this model, one can make the approximation that the dimeric complexes dissociate irreversibly to the monomeric species. In this case, the same mathematical equations apply to the ML*2 model that applied to the ML2 model.
General description
A higher level of modeling, the ML3 model involves four active catalytic complexes: MLRLRLR, MLSLSLS, MLRLRLS, MLSLSLR. Unlike the ML2 model, where only the two homochiral complexes reacted to form enantiomerically enriched product, all four of the catalytic complexes react enantioselectively. However, the same steady state assumption applies to the equilibrium between unbound and bound catalytic complexes as in the more simple ML2 model. This relationship is shown below in Figure 7.

Mathematical modeling
Calculating the eeproduct is considerably more challenging than in the simple ML2 model. Each of the two heterochiral catalytic complexes should react at the same rate. The homochiral catalytic complexes, similar to the ML2 case, should also react at the same rate. As such, the correction parameter g is still calculated as the rate of the heterochiral catalytic complex divided by the rate of the homochiral catalytic complex. However, since the heterochiral complexes lead to enantiomerically enriched product, the overall equation for calculating the eeproduct becomes more difficult. In Figure 8., the mathematical formula for calculating enantioselectivity is shown.

Figure 8: The mathematical formula describing an ML3 system. The eeproduct is calculated by multiplying the eemax by the correction factor developed by Kagan and co-workers.[4]
Interpretation of the ML3 Model
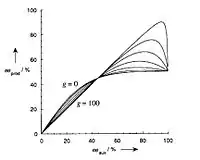
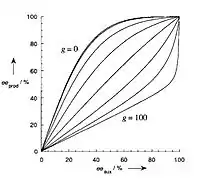
In general, interpreting the correction parameter values of g to predict positive and negative non-linear effects is considerably more difficult. In the case where the heterochiral complexes MLRLRLS and MLSLSLR are less reactive than the homochiral complexes MLSLSLS and MLRLRLR, a kinetic behavior similar to the ML2 model is observed (Figure 9). However, a substantially different behavior is observed in the case where the heterochiral complexes are more reactive than the homochiral complexes.
General Description
Often described adjacent or in collaboration with the ML2 model, the reservoir effect describes the scenario in which part of the chiral ligand is allocated to a pool of inactive heterochiral catalytic complexes outside the catalytic cycle.[4] A pool of unreactive heterochiral catalysts, described with an eepool, develops an equilibrium with the catalytically active homochiral complexes, described with an eeeffective.[8] Depending on the concentration of the inactive pool of catalysts, one can calculate the enantiopurity of the active catalyst complexes. The general result of the reservoir effect is an asymmetric amplification, also known as a (+)-NLE.[3]
Origin of the Reservoir Effect
The pool of unreactive catalytic complexes, as described in the reservoir effect, can be the result of several factors. One of these could potentially be an aggregation effect amongst the heterochiral catalytic complexes that takes place prior to the steady state equilibrium.[3]
Early Examples of the Non-Linear Effect
Sharpless Epoxidation of Geraniol
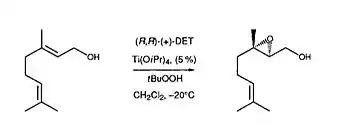
In 1986, Kagan and co-workers were able to demonstrate NLE with the Sharpless epoxidation of (E)-Geraniol (Figure 11). Under Sharpless oxidizing conditions with Ti(O-i-Pr)4/(+)-DET/t-BuOOH, Kagan and coworkers were able to demonstrate that there was a non-linear correlation between the eeproduct and the ee of the chiral catalyst, diethyl tartrate (DET).[3] As one can see from Figure 11, a greater eeproduct than expected was observed. According to the ML2 model, Kagan and coworkers were able to conclude that a less reactive heterochiral DET complex was present. This would therefore explain the asymmetric amplification observed. The NLE data is also consistent with the Sharpless mechanism of asymmetric epoxidation.[10]
Asymmetric Sulfide Oxidation
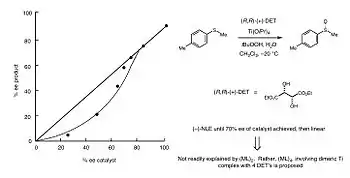
In 1994, Kagan and co-workers reported a NLE in asymmetric sulfide oxidation. The goodness of fit for the reaction data matched the ML4 model. This implied that a dimeric Titanium complexed with 4 DET ligands was the active catalytic species.[3] In this case, the reaction rate would be significantly faster relative to ideal reaction kinetics. The downfall, as is the case in all (−)-NLE scenarios, is that the enantioselectivity was lower than expected.[3] Below, in Figure 12, one can see the concavity of the data points is highly indicative of a (−)-NLE.[1]
Prebiotic Catalysis and the Non-linear Effect
In pre-biotic chemistry, autocatalytic systems play a significant rule in understanding the origin of chirality in life.[6] An autocatalytic reaction, a reaction in which the product acts as a catalyst for itself, serves as a model for homochirality. The asymmetric Soai reaction is commonly referred to as chemical plausibility for this pre-biotic hypothesis. In this system, an asymmetric amplification is observed during the process of autocatalytic catalysis. Professor Donna Blackmond has studied the NLE of this reaction extensively using Kagan's ML2 model. From this mathematical analysis, Blackmond was able to conclude that a dimeric, homochiral complex was the active catalyst in promoting homochirality for the Soai reaction.[3][6]
Notes
- Guillaneux, Denis; Zhao, Shu-Hai; Samuel, Odile; Rainford, David; Kagan, Henri B. (October 1994). "Nonlinear Effects in Asymmetric Catalysis". Journal of the American Chemical Society. 116 (21): 9430–9439. doi:10.1021/ja00100a004.
- Satyanarayana, Tummanapalli; Abraham, Susan; Kagan, Henri B. (5 January 2009). "Nonlinear Effects in Asymmetric Catalysis". Angewandte Chemie International Edition. 48 (3): 456–494. doi:10.1002/anie.200705241.
- Blackmond, Donna G. (December 1997). "Mathematical Models of Nonlinear Effects in Asymmetric Catalysis: New Insights Based on the Role of Reaction Rate". Journal of the American Chemical Society. 119 (52): 12934–12939. doi:10.1021/ja973049m.
- Girard, Christian; Kagan, Henri B. (16 November 1998). "Nonlinear Effects in Asymmetric Synthesis and Stereoselective Reactions: Ten Years of Investigation". Angewandte Chemie International Edition. 37 (21): 2922–2959. doi:10.1002/(SICI)1521-3773(19981116)37:21<2922::AID-ANIE2922>3.0.CO;2-1.
- Wynberg, Hans; Feringa, Ben (January 1976). "Enantiomeric recognition and interactions" (PDF). Tetrahedron. 32 (22): 2831–2834. doi:10.1016/0040-4020(76)80131-7.
- Blackmond, D. G. (5 April 2004). "Asymmetric Catalysis Special Feature Part II: Asymmetric autocatalysis and its implications for the origin of homochirality". Proceedings of the National Academy of Sciences. 101 (16): 5732–5736. Bibcode:2004PNAS..101.5732B. doi:10.1073/pnas.0308363101. PMC 395976. PMID 15067112.
- Puchot, C.; Samuel, O.; Dunach, E.; Zhao, S.; Agami, C.; Kagan, H. B. (April 1986). "Nonlinear effects in asymmetric synthesis. Examples in asymmetric oxidations and aldolization reactions". Journal of the American Chemical Society. 108 (9): 2353–2357. doi:10.1021/ja00269a036.
- Blackmond, Donna G. (June 2000). "Kinetic Aspects of Nonlinear Effects in Asymmetric Catalysis". Accounts of Chemical Research. 33 (6): 402–411. doi:10.1021/ar990083s.
- Kagan, Henri B. (2001). "Nonlinear Effects in Asymmetric Catalysis: A Personal Account". Synlett. 2001 (Special Issue): 0888–0899. doi:10.1055/s-2001-14660.
- Finn, M. G.; Sharpless, K. Barry (January 1991). "Mechanism of asymmetric epoxidation. 2. Catalyst structure". Journal of the American Chemical Society. 113 (1): 113–126. doi:10.1021/ja00001a019.