Singleton Merten syndrome
Singleton Merten Syndrome is an autosomal dominant genetic disorder with variable expression with an onset of symptoms during childhood.
| Singleton Merten syndrome | |
|---|---|
| Other names | Singleton-Merten dysplasia |
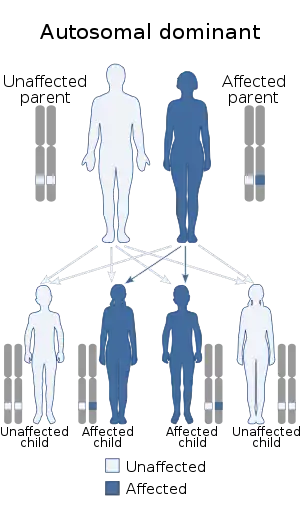 | |
| Singleton Merten syndrome is inherited in an autosomal dominant manner | |
Signs and symptoms
The patients often present with a history of fever of unknown origin, muscular weakness, poor development, abnormal dentition, normal serum calcium, phosphorus, and alkaline phosphatase levels. Associated clinical findings also include glaucoma, photosensitivity, heart block, foot deformities, and chronic psoriasiform skin lesions.
Genetics
This condition has been associated with mutations in the retinoic acid-inducible gene I (DDX58) and melanoma differentiation-associated protein 5 (IFIH1) genes.[1]
Diagnosis
Radiological findings
The classic radiologic findings were first described by Edward B. Singleton and David Merten in 1973.
Typical radiographic appearances include skeletal demineralization, expanded shafts of the metacarpals and phalanges with widenend medullary cavities, cardiomegaly, and intramural calcification of the proximal aorta with occasional extension into the aortic or mitral valves.
Other commonly seen radiographic findings include shallow acetabular fossa, subluxation of the femoral head, coxa valga, hypoplastic radial epiphysis, soft tissue calcifications between the radius and ulna, constriction of the proximal radial shaft, acro-osteolysis, and equinovarus foot deformities.
Sources
References
- Ferreira CR, Crow YJ, Gahl WA, Gardner PJ, Goldbach-Mansky R, Hur S, de Jesús AA, Nehrebecky M, Park JW, Briggs TA (2018) DDX58 and classic Singleton-Merten syndrome. J Clin Immunol
| Classification | |
|---|---|
| External resources |