Strømme syndrome
Strømme syndrome is a very rare autosomal recessive genetic condition characterised by intestinal atresia (in which part of the intestine is missing), eye abnormalities and microcephaly. The intestinal atresia is of the "apple-peel" type, in which the remaining intestine is twisted around its main artery. The front third of the eye is typically underdeveloped, and there is usually moderate developmental delay. Less common features include an atrial septal defect, increased muscle tone or skeletal abnormalities.[2][3] Physical features may include short stature, large, low-set ears, a small jaw, a large mouth, epicanthic folds or fine, sparse hair.[2][3][5]
| Strømme syndrome | |
|---|---|
| Other names | Stromme syndrome, apple-peel intestinal atresia–ocular anomalies–microcephaly syndrome,[1] jejunal atresia–microcephaly–ocular anomalies syndrome,[1] apple peel syndrome with microcephaly and ocular anomalies,[2] jejunal atresia with microcephaly and ocular anomalies,[2] (formerly) primary ciliary dyskinesia 31 (CILD31)[2] |
 | |
| Female infant with Strømme syndrome showing microcephaly | |
| Pronunciation |
|
| Specialty | Medical genetics |
| Symptoms | Apple-peel intestinal atresia, underdeveloped eyes, microcephaly with developmental delay; sometimes additional symptoms or fewer symptoms[2][3] |
| Causes | Genetic (autosomal recessive mutation in CENPF)[2][3] |
| Diagnostic method | Based on symptoms, genetic testing[4] |
| Prognosis | Not yet certain. Good for most, though perinatal mortality possible in the most severe cases.[3][4] |
| Frequency | Not yet known. Around 13 individuals diagnosed as of 2017.[2] |
The syndrome is caused by mutations in both copies of the CENPF gene, which codes for centromere protein F.[2][3] This protein is involved in cell division, in which it forms part of a disc-shaped protein complex known as a kinetochore. CENPF also has a role in orienting long, cylindrical structures called microtubules to form thin cell protrusions called cilia, which send and receive signals to trigger cell division, migration or differentiation. Mutations in the gene result in slower cell division and some embryonic developmental processes being disrupted or not completed, and the syndrome can be classified as a ciliopathy.[2][6][7] The syndrome is typically diagnosed based on the symptoms, but genetic testing provides a full confirmation.[4][7]
Treatment centres around the symptoms. The intestinal atresia is usually surgically correctable in infancy with anastomosis.[3] The prognosis is not yet certain. Those who have survived birth and infancy (the majority) have continued to live through childhood and adolescence, but a large minority with the most severe cases have died before or shortly after birth.[2][3][4]
The prevalence is not yet known. As of 2017, around 13 individuals had been diagnosed.[2] The syndrome was first identified based on symptoms in two siblings by Norwegian paediatrician Petter Strømme and his associates in 1993.[2][8] It was named after him in a 2008 study describing another patient.[2][9] In 2015, mutations in CENPF were first identified as pathogenic,[2][6] and a 2016 genetic analysis of Strømme's original two siblings found that both had mutations in both of their copies of CENPF, establishing it as the cause of the syndrome.[2][7]
Signs and symptoms
The most common symptoms of Strømme syndrome are intestinal atresia, eye abnormalities and microcephaly. However, the syndrome has a wide range of severity that generally runs in the family it presents in, ranging from only mild learning disability and microcephaly with no other features in some families[2][6] to death in utero with severe kidney, heart, eye, skeletal, brain and intestinal malformations in others.[2][6][7] The variable severity is usually due to the presence or absence in each family of mutations in other genes with similar functions to CENPF.[10]
Intestinal
Individuals with Strømme syndrome are typically born with intestinal atresia, in which parts of the intestine are narrow or missing, leading to neonatal bowel obstruction that must be operated on.[3] The intestinal atresia is of the "apple-peel" type, an uncommon type in which the remaining portion of the intestine is found twisted around its main artery, and this usually affects the jejunum.[2][3] Often, much of the bowel is missing in this form of atresia.[9] There can sometimes also be intestinal malrotation.[2][3][8][11]
At least two individuals with the syndrome in literature have avoided intestinal atresia, one of which had a sibling with the same mutations who did not.[6][10] In two siblings who did not survive to term, the intestinal atresia (which also included duodenal atresia) and malrotation were more severe.[2][7]
Eyes
The eyes are often smaller and underdeveloped, usually more severely in one eye than the other.[2] This can manifest as a coloboma (hole) in the iris, cataracts, opacity of the cornea (leukoma), sclerocornea (in which the white of the eye blends into the cornea), a small cornea (microcornea) and synechia (in which the iris adheres to the cornea or lens).[3] This underdevelopment of the front of the eye, known as anterior segment dysgenesis (which includes Peters' anomaly), can lead to an increased risk of glaucoma from high intraocular pressure, due to impaired eye fluid drainage, though this hadn't been reported in any of the affected individuals as of 2017.[3]
There may also be crossing of the eyes (esotropia),[8] and less commonly there may be twisted retinal blood vessels or optic nerve hypoplasia.[3][7] The eye anomalies can result in an inability to focus (astigmatism) as well as amblyopia, in which the brain begins to fail to process input from the weaker eye during childhood.[7]
Neurological
Those affected with the syndrome usually have microcephaly.[2][3] A large minority also have pachygyria (fewer ridges in the brain) or lissencephaly (shallower ridges).[9][10][12] Developmental delay is usually present. It has usually been moderate-to-severe, but in some cases it has been mild.[3] A few of those affected have had increased muscle tone.[2][5][11] One individual had cortical heterotopia, which is a sign of impaired neuronal cell migration during neural development.[13]
Agenesis or hypoplasia of the corpus callosum and cerebellum have been found in at least one living affected individual and several who did not survive to term.[2][6][12] Hydrocephalus occurred in one living individual 9 months after birth[2][5] and in four who did not survive to term.[2][6] Hydrocephalus was also observed in zebrafish whose CENPF genes were experimentally knocked out.[2][6] Cerebellar hypoplasia in association with hydrocephalus is called Dandy–Walker malformation and is found in a number of other ciliopathies, sometimes together with agenesis of the corpus callosum.[14]
Physical features
Physical features are variable but usually include short stature, large, low-set ears, a high nasal bridge, a small jaw and a large mouth.[2][6][10] Some of those affected have had epicanthal folds or fine, sparse hair.[5] One individual was born with a skin tag on the left cheek.[2][10] Four affected individuals who did not survive to term had cleft palate.[2][6]
Heart
A minority of those affected have been born with an atrial septal defect, a type of congenital heart defect.[2][10] One affected individual had a ventricular septal defect and neonatal peripheral oedema in the legs.[12] Two individuals who did not survive to term had a patent foramen ovale, a specific type of atrial septal defect, as well as reduced heart muscle tissue (myocardium) and abnormally small heart muscle cells (cardiomyocytes).[2][7]
Skeletal
One affected individual had hip dysplasia, leading to dislocation,[5] and another had metopic craniosynostosis, leading to a metopic ridge.[2][10] Two individuals who did not survive to term had polydactyly of the thumb (preaxial polydactyly), flattened vertebrae (platyspondyly) and a rare chest wall malformation called a sternal cleft.[2][7]
Kidneys
One living individual had underdeveloped and malformed kidneys,[12] and two siblings who did not survive to term had underdeveloped kidneys and ureters, leading to a build-up of urine called hydronephrosis.[2][7]
Blood
One affected person had a reduced number of platelets (thrombocytopaenia) in infancy, requiring transfusion. Platelets are cellular fragments formed from protrusions on megakaryocytes that enable blood clotting. Blood symptoms have not yet been reported in any other affected individuals.[12]
Cause
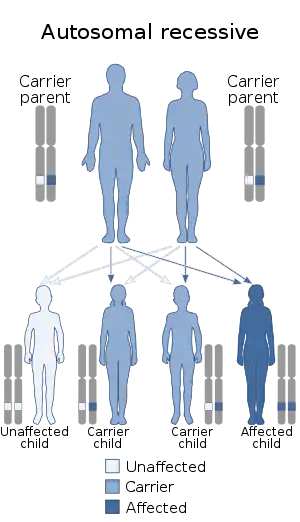
Strømme syndrome is caused by mutations in both copies of the CENPF gene, located on the long arm of chromosome 1.[2][3] CENPF codes for centromere protein F. Centromere proteins are involved in the separation of chromosomes during cell division. This is through forming part of kinetochores, which are disc-shaped protein complexes that allow the centromeres of chromosomes (in the dividing form, known as chromatids) to attach to microtubules in the cell (forming what is called the spindle apparatus). This allows the microtubules to pull the chromosomes apart in the process of dividing the cell. Mutations in this gene lead to impaired cell division during early development. Mitosis has been found to take longer when CENPF is mutated.[6][7]
Microtubules are protein structures that are part of the cytoskeleton and are necessary for cells to have diverse, complex shapes and migratory ability. They are made by the centrosome, which contains a pair of cylindrical centrioles at right-angles to each other. Before division, CENPF localises at the end of one of the centrioles (the mother centriole) in order to orient microtubules correctly to form thin cellular projections called cilia. Most cilia are primary cilia, which are involved in cell signalling, sending and receiving signals to trigger cell migration, division or differentiation. Mutations in CENPF disrupt this ability to form cilia; cilia have been found to be fewer in number and shorter when CENPF is mutated. Strømme syndrome therefore falls under the classification of diseases known as ciliopathies.[2][6]
Mutations that have been identified in CENPF have been mostly nonsense mutations, which result in the protein being cut short and usually non-functional as a result, but frameshift and splice-site mutations have also been identified. Several of the nonsense mutations that have led to this syndrome have been in exon 12 of the gene (out of 20), but mutations in other exons have been identified.[6][7] Severity and symptoms of the syndrome have been variable regardless of the type of mutation but generally consistent within families, suggesting the severity may depend on the presence of mutations in other genes that perform similar functions to or otherwise interact with or affect CENPF (a phenomenon known as epistasis).[10] It has been suggested that an interaction between CENPF and NDE1, which causes microlissencephaly when mutated, is related to the microcephaly in Strømme syndrome.[6][10]
Diagnosis
Diagnosis is typically achieved by observation of symptoms; however, genetic testing provides a full confirmation. The microcephaly, intestinal atresia and some of the eye abnormalities are observable on prenatal ultrasound.[2][4] Brain MRI scans can reveal any brain anomalies that could be associated with the syndrome.[4] Methods of genetic detection include whole exome sequencing and panel testing, which involves sequencing a selection of potential genes involved.[2][4][10] Sanger sequencing can confirm the nature of the mutation.[2][10]
Once a family has been identified as being carriers for mutated CENPF genes, prenatal diagnosis and preimplantation genetic diagnosis can be offered for future conceptions.[4]
Treatment
Treatment targets the symptoms. The intestinal atresia is usually surgically correctable in infancy with anastomosis; however, no eye surgery had been reported as of 2017.[3] Van Bever et al. recommended monitoring patients for glaucoma.[9]
Prognosis
The prognosis is not yet certain. The majority of those affected have survived birth and infancy, and these individuals have continued to live through childhood and adolescence. However, a large minority with the most severe presentations have died before birth or shortly after.[4][6][7] The oldest known people with the syndrome, Strømme's original two siblings, who had a mild-to-moderate presentation, were in their twenties and in employment as of 2016.[2][7]
Epidemiology
The prevalence of the syndrome is not yet known. As of 2017, around 13 individuals had been diagnosed.[2]
History
The condition was first identified in 1993, when Norwegian paediatrician Petter Strømme and his associates observed two infant siblings with microcephaly and eye abnormalities alongside apple-peel intestinal atresia at Rogaland Central Hospital in Stavanger, Norway. They proposed that it constituted a new syndrome.[2][8] Later studies by Slee and Goldblatt (1996),[5] Shanske et al. (2002),[13] Bellini et al. (2002)[11] and others observed other patients with similar symptoms who appeared to have the syndrome.[2] In 2008, Van Bever et al. proposed that the syndrome be named after Strømme, after encountering another patient who seemed to have the syndrome.[2][9]
In 2015, Waters et al. conducted a genetic analysis on a British family in which four foetuses had miscarried with symptoms of a ciliopathy. They found that the foetuses had mutations in both copies of CENPF. They subsequently analysed a cohort of 1,000 individuals with microcephaly and found that one of them, a girl, had mutations in both of her copies of CENPF. Her learning delay was mild-to-moderate, and she did not have any other issues with her bodily systems. This confirmed for the first time that mutations in CENPF are pathogenic.[2][6]
In 2016, Filges et al. followed up with Strømme's original two siblings and found using whole exome sequencing that they both had mutations in both of their copies of CENPF, establishing mutations in CENPF as the cause of Strømme syndrome.[2][7]
Notable cases
- In May 2017, Ruby Ardolf (born November 11, 2004), from Minnesota, United States and diagnosed with Strømme syndrome, appeared in an Instagram video answering questions from her mother Angela which went viral, gaining over 500,000 views in a week.[15] Angela manages a website, online store and YouTube channel for her daughter, with over 170,000 subscribers as of July 2020.[16]
See also
References
- "Orphanet: Stromme syndrome". www.orpha.net. Retrieved 8 December 2019.
- "OMIM Entry - # 243605 - STROMME SYNDROME; STROMS". www.omim.org. Retrieved 27 September 2018.
- "Strømme Syndrome | Hereditary Ocular Diseases". disorders.eyes.arizona.edu. Archived from the original on 24 July 2017. Retrieved 27 September 2018.
- Filges, Isabel; Stromme, Petter (2020). "CUGC for Stromme syndrome and CENPF-related disorders". European Journal of Human Genetics. 28 (1): 132–136. doi:10.1038/s41431-019-0498-y. ISSN 1476-5438. PMC 6906375. PMID 31488893.
- Slee, J.; Goldblatt, J. (October 1996). "Further evidence for a syndrome of "apple peel" intestinal atresia, ocular anomalies and microcephaly". Clinical Genetics. 50 (4): 260–262. doi:10.1111/j.1399-0004.1996.tb02640.x. ISSN 0009-9163. PMID 9001813.
- Waters, Aoife M.; Asfahani, Rowan; Carroll, Paula; Bicknell, Louise; Lescai, Francesco; Bright, Alison; Chanudet, Estelle; Brooks, Anthony; Christou-Savina, Sonja; Osman, Guled; Walsh, Patrick (March 2015). "The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes". Journal of Medical Genetics. 52 (3): 147–156. doi:10.1136/jmedgenet-2014-102691. ISSN 1468-6244. PMC 4345935. PMID 25564561.
- Filges, Isabel; Bruder, Elisabeth; Brandal, Kristin; Meier, Stephanie; Undlien, Dag Erik; Waage, Trine Rygvold; Hoesli, Irene; Schubach, Max; de Beer, Tjaart; Sheng, Ying; Hoeller, Sylvia (April 2016). "Strømme Syndrome Is a Ciliary Disorder Caused by Mutations in CENPF". Human Mutation. 37 (4): 359–363. doi:10.1002/humu.22960. ISSN 1098-1004. PMID 26820108.
- Strømme, P.; Dahl, E.; Flage, T.; Stene-Johansen, H. (October 1993). "Apple peel intestinal atresia in siblings with ocular anomalies and microcephaly". Clinical Genetics. 44 (4): 208–210. doi:10.1111/j.1399-0004.1993.tb03881.x. ISSN 0009-9163. PMID 8261651.
- van Bever, Yolande; van Hest, Liselotte; Wolfs, Roger; Tibboel, Dick; van den Hoonaard, Thelma L.; Gischler, Saskia J. (15 February 2008). "Exclusion of a PAX6, FOXC1, PITX2, and MYCN mutation in another patient with apple peel intestinal atresia, ocular anomalies and microcephaly and review of the literature". American Journal of Medical Genetics. Part A. 146A (4): 500–504. doi:10.1002/ajmg.a.32169. ISSN 1552-4833. PMID 18203155.
- Ozkinay, Ferda; Atik, Tahir; Isik, Esra; Gormez, Zeliha; Sagiroglu, Mahmut; Sahin, Ozlem Atan; Corduk, Nergul; Onay, Huseyin (June 2017). "A further family of Stromme syndrome carrying CENPF mutation". American Journal of Medical Genetics. Part A. 173 (6): 1668–1672. doi:10.1002/ajmg.a.38173. ISSN 1552-4833. PMID 28407396.
- Bellini, Carlo; Mazzella, Massimo; Arioni, Cesare; Fondelli, Maria Paola; Serra, Giovanni (15 June 2002). ""Apple-peel" intestinal atresia, ocular anomalies, and microcephaly syndrome: brain magnetic resonance imaging study". American Journal of Medical Genetics. 110 (2): 176–178. doi:10.1002/ajmg.10392. ISSN 0148-7299. PMID 12116257.
- Dorum, Bayram Ali; Şambel, Irmak Tanal; Özkan, Hilal; Kırıştıoğlu, İrfan; Köksal, Nilgün (21 March 2017). "Stromme Syndrome: New Clinical Features". APSP Journal of Case Reports. 8 (2): 14. doi:10.21699/ajcr.v8i2.564. ISSN 2218-8185. PMC 5371687. PMID 28401041.
- Shanske, Alan L.; Gurland, Judith E.; Mbekeani, Joyce N.; Bello, Jacqueline A.; Campbell, Deborah; Kleinhaus, Sylvain (January 2002). "Possible new syndrome of microcephaly with cortical migration defects, Peters anomaly and multiple intestinal atresias: a multiple vascular disruption syndrome". Clinical Dysmorphology. 11 (1): 67–69. doi:10.1097/00019605-200201000-00014. ISSN 0962-8827. PMID 11822709.
- Badano, Jose L.; Mitsuma, Norimasa; Beales, Phil L.; Katsanis, Nicholas (1 September 2006). "The Ciliopathies: An Emerging Class of Human Genetic Disorders". Annual Review of Genomics and Human Genetics. 7 (1): 125–148. doi:10.1146/annurev.genom.7.080505.115610. ISSN 1527-8204. PMID 16722803.
- "Lakeville Mom, Daughter Go Viral on Instagram". CBS Minnesota. 24 May 2017. Retrieved 16 December 2019.
- Chiu, Jessica. "On YouTube, people with disabilities create content to show and normalize their experiences". The Washington Post. Retrieved 16 December 2019.
External links
| Classification | |
|---|---|
| External resources |