Synthesis of nucleosides
Synthesis of nucleosides involves the coupling of a nucleophilic, heterocyclic base with an electrophilic sugar. The silyl-Hilbert-Johnson (or Vorbrüggen) reaction, which employs silylated heterocyclic bases and electrophilic sugar derivatives in the presence of a Lewis acid, is the most common method for forming nucleosides in this manner.[1]
Introduction
Nucleosides are typically synthesized through the coupling of a nucleophilic pyrimidine, purine, or other basic heterocycle with a derivative of ribose or deoxyribose that is electrophilic at the anomeric carbon. When an acyl-protected ribose is employed, selective formation of the β-nucleoside (possessing the S configuration at the anomeric carbon) results from neighboring group participation. Stereoselective synthesis of deoxyribonucleosides directly from deoxyribose derivatives is more difficult to achieve because neighboring group participation cannot take place.
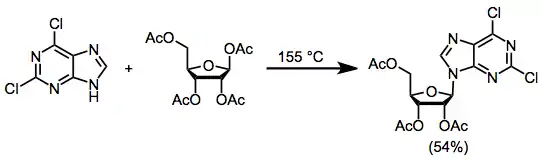
Three general methods have been used to synthesize nucleosides from nucleophilic bases and electrophilic sugars. The fusion method involves heating the base and acetyl-protected 1-acetoxyribose to 155 °C and results in the formation of the nucleoside with a maximum yield of 70%.[2]
(1)
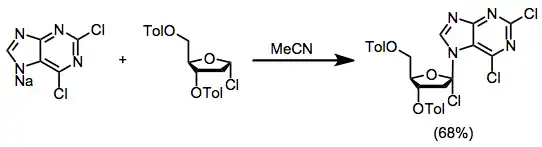
The metal salt method involves the combination of a metal salt of the heterocycle with a protected sugar halide. Silver[3] and mercury[4] salts were originally used; however, more recently developed methods use sodium salts.[5]
(2)
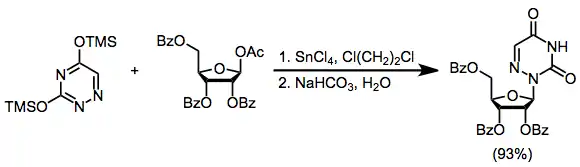
The silyl-Hilbert-Johnson (SHJ) reaction (or Vorbrüggen reaction), the mildest general method for the formation of nucleosides, is the combination of a silylated heterocycle and protected sugar acetate (such as 1-O-acetyl-2,3,5-tri-O-benzoyl-beta-D-ribofuranose) in the presence of a Lewis acid.[6] Problems associated with the insolubility of the heterocyclic bases and their metal salts are avoided; however, site selectivity is sometimes a problem when heterocycles containing multiple basic sites are used, as the reaction is often reversible.
(3)
Mechanism and Stereochemistry
The Silyl-Hilbert-Johnson Reaction
The mechanism of the SHJ reaction begins with the formation of the key cyclic cation 1. Nucleophilic attack at the anomeric position by the most nucleophilic nitrogen (N1) then occurs, yielding the desired β-nucleoside 2.[7] A second reaction of this nucleoside with 1 generates bis(riboside) 3. Depending on the nature of the Lewis acid used, coordination of the nucleophile to the Lewis acid may be significant. Reaction of this "blocked" nucleophile with 1 results in undesired constitutional isomer 4, which may undergo further reaction to 3.[8] Generally Lewis acid coordination is not a problem when a Lewis acid such as trimethylsilyl triflate is used; it is much more important when a stronger Lewis acid like tin(IV) chloride is employed.[7]
(4)
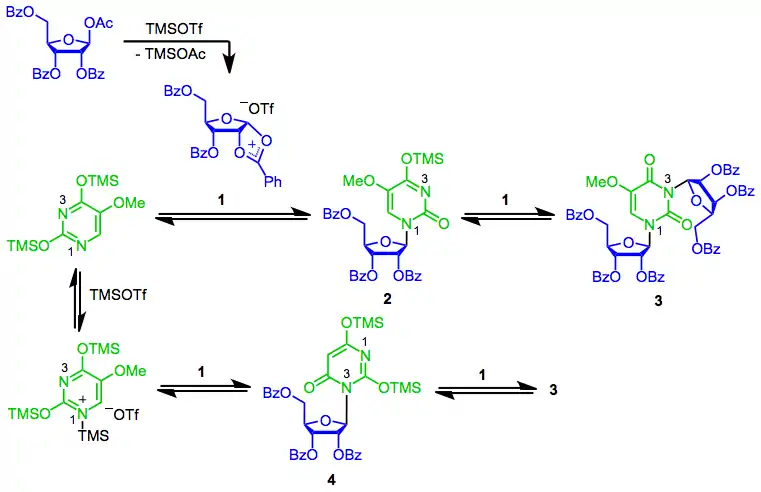
2-Deoxysugars are unable to form the cyclic cation intermediate 1 because of their missing benzoyl group; instead, under Lewis acidic conditions they form a resonance-stabilized oxocarbenium ion. The diastereoselectivity of nucleophilic attack on this intermediate is much lower than the stereoselectivity of attack on cyclic cation 1. Because of this low stereoselectivity, deoxyribonucleosides are usually synthesized using methods other than the SHJ reaction.[9]
Scope and Limitations
The silyl-Hilbert-Johnson reaction is the most commonly used method for the synthesis of nucleosides from heterocyclic and sugar-based starting materials. However, the reaction suffers from some issues that are not associated with other methods, such as unpredictable site selectivity in some cases (see below). This section describes both derivatives of and alternatives to the SHJ reaction that are used for the synthesis of nucleosides.
Silyl-Hilbert-Johnson Reactions
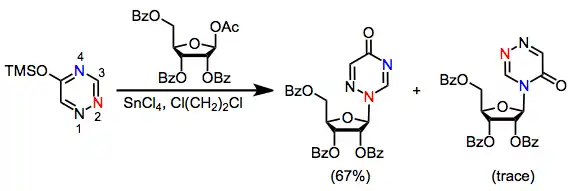
Because most heterocyclic bases contain multiple nucleophilic sites, site selectivity is an important issue in nucleoside synthesis. Purine bases, for instance, react kinetically at N3 and thermodynamically at N1 (see Eq. (4)).[4] Glycosylation of thymine with protected 1-acetoxy ribose produced 60% of the N1 nucleoside and 23% of the N3 nucleoside. Closely related triazines, on the other hand, react with complete selectivity to afford the N2 nucleoside.[10]
(5)
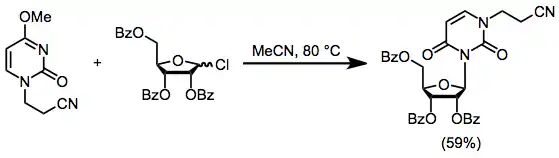
The most nucleophilic nitrogen can be blocked through alkylation prior to nucleoside synthesis. Heating the blocked nucleoside in Eq. (6) in the presence of a protected sugar chloride provides the nucleoside in 59% yield. Reactions of this type are hampered by alkylation of the heterocycle by incipient alkyl chloride.[11]
(6)
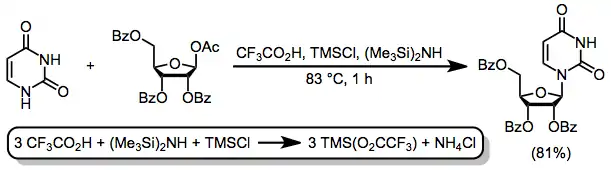
Silylated heterocyclic bases are susceptible to hydrolysis and somewhat difficult to handle as a result; thus, the development of a one-pot, one-step method for silylation and nucleoside synthesis represented a significant advance.[12] The combination of trifluoroacetic acid (TFA), trimethylsilyl chloride (TMSCl), and hexamethyldisilazide (HMDS) generates trimethylsilyl trifluoroacetate in situ, which accomplishes both the silylation of the heterocycle and its subsequent coupling with the sugar.[13]
(7)
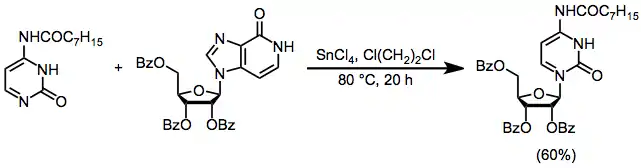
Other Methods for Nucleoside Synthesis
Transglycosylation, which involves the reversible transfer of a sugar moiety from one heterocyclic base to another, is effective for the conversion of pyrimidine nucleosides to purine nucleosides. Most other transglycosylation reactions are low yielding due to a small thermodynamic difference between equilibrating nucleosides.[14]
(8)
Deoxyribose-derived electrophiles are unable to form the cyclic cation 1; as a result, the stereoselective synthesis of deoxyribonucleosides is more difficult than the synthesis of ribonucleosides. One solution to this problem involves the synthesis of a ribonucleoside, followed by protection of the 3'- and 5'-hydroxyl groups, removal of the 2'-hydroxyl group through a Barton deoxygenation, and deprotection.[15]
(9)
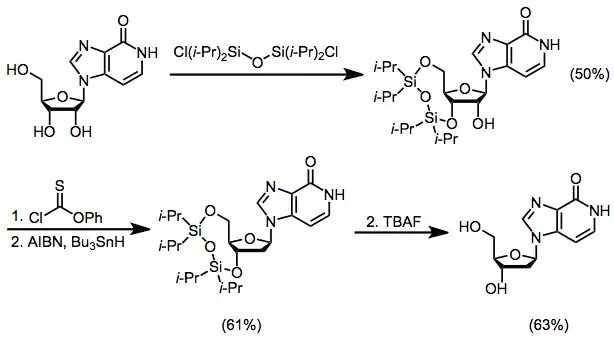
Comparison with Other Methods
A useful alternative to the methods described here that avoids the site selectivity concerns of the SHJ reaction is tandem Michael reaction/cyclization to simultaneously form the heterocyclic base and establish its connection to the sugar moiety.[16]
(10)
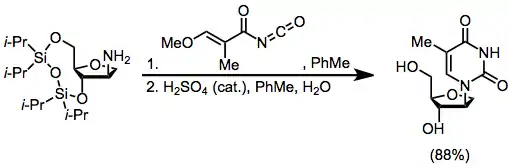
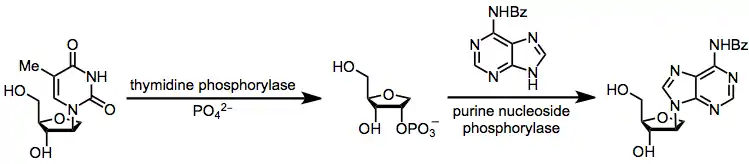
A second alternative is enzymatic transglycosylation, which is completely kinetically controlled (avoiding issues of chemical transglycosylation associated with thermodynamic control). However, operational complications associated with the use of enzymes are a disadvantage of this method.[17]
(11)
Experimental Conditions and Procedure
Typical Conditions
The sugar derivatives used for SHJ reactions should be purified, dried, and powdered before use. Intramolecular Friedel-Crafts reaction of the aromatic ring of a benzoate ester at the 2-position of 1-acetoxy ribose in the presence of a Lewis acid has been observed, and represents a potential side reaction.[18] Heterocycles must not be too basic in order to avoid excessive complexation with the Lewis acid; amino-substituted heterocycles such as cytosine, adenine, and guanine react slowly or not at all under SHJ conditions (although their N-acetylated derivatives react more rapidly).
Silylation is most commonly accomplished using HMDS, which evolves ammonia as the only byproduct of silylation. Catalytic or stoichiometric[19] amounts of acidic additives such as trimethylsilyl chloride accelerate silylation; when such an additive is used, ammonium salts will appear in the reaction as a turbid impurity.
Lewis acids should be distilled immediately before use for best results. More than about 1.2-1.4 equivalents of Lewis acid are rarely needed. Acetonitrile is the most common solvent employed for these reactions, although other polar solvents are also common. Workup of reactions employing TMSOTf involves treatment with an ice-cold solution of sodium bicarbonate and extraction of the resulting sodium salts. When tin(IV) chloride is used in 1,2-dichloroethane, workup involves the addition of pyridine and filtering of the resulting pyridine-tin complex, followed by extraction with aqueous sodium bicarbonate.[20]
Example Procedure[21]
(12)
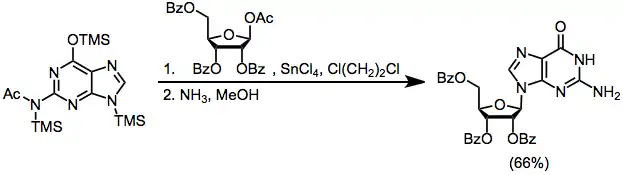
To a stirred mixture of 13.5 mL (4.09 mmol) of a 0.303 N standard solution of silylated N2-acetylguanine in 1,2-dichloroethane and 1.86 g (3.7 mmol) of benzoate-protected 1-acetoxy ribose in 35 mL of 1,2-dichloroethane was added 6.32 mL (4.46 mmol) of a 0.705 N standard solution of TMSOTf in 1,2-dichloroethane. The reaction mixture was heated at reflux for 1.5–4 hours, and then diluted with CH2Cl2. On workup with ice-cold NaHCO3 solution, there was obtained 2.32 g of crude product, which was kept for 42 hours in 125 mL of methanolic ammonia at 24°. After workup, recrystallization from H2O gave, in two crops, 0.69 g (66%) of pure guanosine, which was homogeneous (Rf 0.3) in the partition system n-butanol:acetic acid:H2O (5:1:4) and whose 1H NMR spectrum at 400 MHz in D2O showed only traces of the undesired N7-anomer of guanosine. 1H NMR (CDCl3): δ 3.55, 3.63, 3.90, 4.11, 4.43, 5.10, 5.20, 5.45, 5.72, 6.52, 7.97, 10.75.
References
- Vorbrüggen, H.; Ruh-Polenz, C. Org. React. 1999, 55, 1. doi:10.1002/0471264180.or055.01
- Diekmann, E.; Friedrich, K.; Fritz, H.-G. J. Prakt. Chem. 1993, 335, 415.
- Fischer, E.; Helferich, B. Chem. Ber. 1914, 47, 210.
- Miyaki, M.; Shimizu, B. Chem. Pharm. Bull. 1970, 18, 1446.
- Kazimierczuk, Z.; Cottam, H. B.; Revankar, G. R.; Robins, R. K. J. Am. Chem. Soc. 1984, 106, 6379.
- Wittenburg, E. Z. Chem. 1964, 4, 303.
- Choi, W-B.; Wilson, L. J.; Yeola, S.; Liotta, D. C.; Schinazi, R. F. J. Am. Chem. Soc. 1991, 113, 9377.
- Vorbrüggen, H.; Niedballa, U.; Krolikiewicz, K.; Bennua, B.; Höfle, G. In Chemistry and Biology of Nucleosides and Nucleotides; Harmon, R. E., Robins, R. K., Townsend, L. B., Eds.; Academic: New York, 1978; p. 251.
- Prystas, M.; Šorm, F. ChemPlusChem 1964, 29, 121.
- Niedballa, U.; Vorbrüggen, H. J. Org. Chem. 1974, 39, 3668.
- Itoh, T.; Melik-Ohanjanian, R. G.; Ishikawa, I.; Kawahara, N.; Mizuno, Y.; Honma, Y.; Hozumi, M.; Ogura, H. Chem. Pharm. Bull. 1989, 37, 3184.
- Vorbrüggen, H.; Bennua, B. Tetrahedron Lett. 1978, 1339.
- Vorbrüggen, H.; Bennua, B. Chem. Ber. 1981, 114, 1279.
- Sugiura, Y.; Furuya, S.; Furukawa, Y. Chem. Pharm. Bull. 1988, 36, 3253.
- Kawasaki, A. M.; Wotring, L. L.; Townsend, L. B. J. Med. Chem. 1990, 33, 3170.
- Nair, V.; Purdy, D. F. Heterocycles 1993, 36, 421.
- Hanrahan, J. R.; Hutchinson, D. W. J. Biotechnol. 1992, 23, 193.
- Martin, O. R. Tetrahedron Lett. 1985, 26, 2055.
- Langer, S. H.; Connell, S.; Wender, I. J. Org. Chem. 1958, 23, 50.
- Patil, V. D.; Wise, D. S.; Townsend, L. B. J. Chem. Soc., Perkin Trans. 1 1980, 1853.
- Vorbrüggen, H.; Krolikiewicz, K.; Bennua, B. Chem. Ber. 1981, 114, 1234.