X-linked reticulate pigmentary disorder
X-linked reticulate pigmentary disorder is a rare X-linked genetic condition in which males manifest multiple systemic symptoms and a reticulated mottled brown pigmentation of the skin, which, on biopsy, demonstrated dermal deposits of amyloid. Females usually only have linear streaks of hyperpigmentation.[1]
| X-linked reticulate pigmentary disorder | |
|---|---|
| Other names | Familial cutaneous amyloidosis,[1] Partington amyloidosis,[1] Partington cutaneous amyloidosis,[1] Partington syndrome type II,[1] reticulate pigmentary disorder,[1] X-linked reticulate pigmentary disorder with systemic manifestations[1] |
.svg.png.webp) | |
| This condition is inherited in an X-linked recessive manner. Carrier females usually only have linear streaks of hyperpigmentation. | |
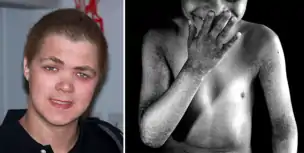
The syndrome is also referred to by the acronym X-Linked-PDR or XLPRD.[2] It's a very rare disease, genetically determined, with a chronic course.
It was characterized in 1981.[3] Mutation of the POLA1 gene leads to loss of expression of the catalytic subunit of DNA polymerase-α and is responsible for XLPDR.[2] Loss of POLA1 expression results in reduced levels of RNA:DNA hybrids in the cytosol and unexpectedly triggers aberrant immune responses (e.g. type I interferon production) which at least in part can account for the symptoms associated with XLPDR.[2] Another trigger of the immunodeficiency phenotype is a functional deficiency of NK cells, major players of innate antiviral immune system.[4]
Presentation
Affected males develop generalized reticular hyperpigmentation in early childhood. Hair often looks bedraggled or brushed backward, hanging low on the forehead. Under XLPDR conditions, autoimmune manifestations are developed due to chronically activated anti-viral type I interferon response, connecting XLPDR with disorders like Aicardi-Goutiere syndrome, Systemic Lupus Erythematosus, Psoriasis, etc. 3 Meanwhile, another typical symptom - immunodeficiency - can be developed due to discovering a functional defect in the cytolytic activity of NK cells. Starokadomskyy at al. discovered that POLA1 deficiency is associated with decreased direct cytotoxicity of NK cells due to disturbances in vesicular traffic. Meanwhile, antibody-dependent cell cytotoxicity (ADCC) remains unchanged in XLPDR NK cells.[4]
The most common manifestations of XLPDR:
- Recurrent respiratory infections
- Dyskeratosis corneal
- Photophobia
- Hypohidrosis (lack of sweat glands)
- NK cell functional deficiency
- Growth retardation
- Gastrointestinal disorders
- Kidney disease
- Kidney stones
- Urinary infections
- Webbed feet or hands
- Electrolyte imbalance
- Retinitis pigmentosa
- Lymphoedema
- Thyroid abnormalities
Not every patient shows all of the listed symptoms. However, skin pathologies, recurrent lung infection, high titer of interferon type I in the blood, and impaired direct cytotoxicity of NK cells are the most common symptoms. In females the disease is characterized by skin rashes linear hyper pigmentation following the Blaschko's lines, morphologically similar to stage 3 pigment incontinence. There are no systemic manifestations associated with XLPDR in females.
See also
- Waardenburg syndrome
- List of cutaneous conditions
References
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. p. 630. ISBN 1-4160-2999-0.
- Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, Pokatayev V, Dozmorov I, Khan S, Miyata N, Fraile G, Raj P, Xu Z, Xu Z, Ma L, Lin Z, Wang H, Yang Y, Ben-Amitai D, Orenstein N, Mussaffi H, Baselga E, Tadini G, Grunebaum E, Sarajlija A, Krzewski K, Wakeland EK, Yan N, de la Morena MT, Zinn AR, Burstein E (2016). "DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis". Nature Immunology. 17: 495–504. doi:10.1038/ni.3409. PMC 4836962. PMID 27019227.CS1 maint: uses authors parameter (link)
- Partington MW, Marriott PJ, Prentice RS, Cavaglia A, Simpson NE (1981). "Familial cutaneous amyloidosis with systemic manifestations in males". Am. J. Med. Genet. 10 (1): 65–75. doi:10.1002/ajmg.1320100109. PMID 6794369.
- Starokadomskyy, Petro; Wilton, Katelynn M.; Krzewski, Konrad; Lopez, Adam; Sifuentes-Dominguez, Luis; Overlee, Brittany; Chen, Qing; Ray, Ann; Gil-Krzewska, Aleksandra; Peterson, Mary; Kinch, Lisa N. (2019-11-01). "NK cell defects in X-linked pigmentary reticulate disorder". JCI Insight. 4 (21). doi:10.1172/jci.insight.125688. ISSN 0021-9738.
External links
| Classification | |
|---|---|
| External resources |