Alpha-mannosidosis
Alpha-mannosidosis is a lysosomal storage disorder,[1] first described by Swedish physician Okerman in 1967.[2] In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency.[2][3][4] Consequently, if both parents are carriers, there will be a 25% chance with each pregnancy that the defective gene from both parents will be inherited, and the child will suffer from the disease. There is a two in three chance that unaffected siblings will be carriers (Figure 1).[4] In livestock alpha-mannosidosis is caused by chronic poisoning with swainsonine from locoweed.
| Alpha-mannosidosis | |
|---|---|
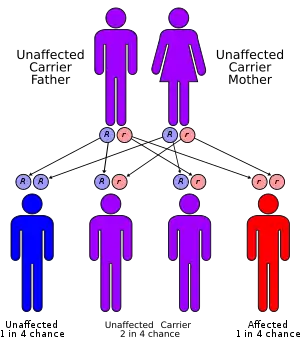 | |
| Alpha-mannosidosis has an autosomal recessive pattern of inheritance Figure 1 | |
| Specialty | Endocrinology |
Symptoms
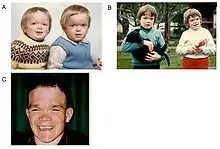
Alpha-mannosidosis is a lifelong multi-systemic progressive disease, with neuromuscular and skeletal deterioration over decades.[2] The timing of the appearance of symptoms correlates with the severity of the disease. The onset of the most severe form of the disease occurs within the first months of life and involves skeletal abnormalities and intellectual disability, with rapid progression leading to death from primary central nervous system involvement or myopathy.[2] However, most neonates with lysosomal storage disorders are asymptomatic and only rarely severely affected.[1][5] This delays diagnosis, particularly as milder forms of the disease involve only mild to moderate intellectual disability, which progresses gradually during childhood or adolescence.[6]
The first decade of life is characterised by the development of hearing impairment, psychomotor delay, recurrent infections, especially upper airway infections, pulmonary infections and acute/serous otitis media infections.[7] Significant changes in a number of facial features may occur, such as: protruding forehead; flattened nasal bridge; small nose; wide mouth; and widely spaced teeth.[2] Muscular weakness or spinal abnormalities can occur due to the build-up of storage materials in the muscle.[2]
Pathophysiology
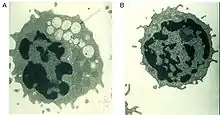
A defective alpha-mannosidase enzyme, which normally helps to break down complex sugars derived from glycoproteins in the lysosome, causes progressive lysosomal accumulation of mannose-rich oligosaccharides in all tissues, resulting in impaired cellular function and apoptosis (Figure 2).[2][8] Complete absence of functionality in this enzyme leads to death during early childhood due to deterioration of the central nervous system.[8] Enzymes with low residual activity lead to a milder form of the disease, with symptoms such as impaired hearing, cognitive impairment, susceptibility to bacterial infections, and skeletal deformities. The course of the disease is progressive.[2][8]
Depending on the severity of the disease, alpha-mannosidosis has been classified into three proposed subtypes, based on severity and age of onset.[2]
- Type 1: A mild form, recognized after age ten years, with absence of skeletal abnormalities, muscle problems (myopathy), and slow progression
- Type 2: A moderate form, recognized before age ten years, with presence of skeletal abnormalities, myopathy, and slow progression. This is the most common form
- Type 3: A severe form, leading to early death from progressive central nervous system involvement
However, given the variety of mutations that have been documented, and the broad range and severity of symptoms, the disease is considered clinically as a continuum.[8][7]
Diagnosis
Alpha Mannosidosis is a progressive disorder, and its presence should be suspected in patients with cognitive disabilities, skeletal changes (e.g. swollen joints, curved spine), hearing loss and recurrent infections. Although children with the condition are often born seemingly normal, their condition deteriorates with age. Alpha-mannosidosis can impact on a patient's quality of life in many ways, including their ability to live independently, socialise or find employment.[2][7]
Generally, phenotypes of alpha-mannosidosis patients are not clearly distinguishable, which makes a prediction of the clinical course for an individual patient challenging.[2] Patients may present to doctors, nurses or health visitors at different stages of progression, and with different ad hoc symptoms, making the link to suspect a diagnosis of alpha-mannosidosis difficult.[2] The main symptoms can also be shared with those of other lysosomal storage disorders, such as mucopolysaccharidosis.[2]
Given the progressive nature of the disease, the earlier a correct diagnosis is achieved the better.[2] The condition is often diagnosed and treated using a multi-disciplinary approach, involving paediatricians, orthopaedics, ophthalmologists, otologists, neurologists, immunologists, neurosurgeons and physiotherapists.[7]
A diagnosis of alpha-mannosidosis is suspected based upon identification of characteristic findings of a multi-symptomatic presentation, a thorough clinical evaluation, a detailed patient history, and results from the diagnostic tests described below:
A. Oligosaccharides in urine
A preliminary investigation may be performed to measure mannose-rich oligosaccharide concentrations in urine. Elevated urinary excretion of mannose-rich oligosaccharides is suggestive, but not diagnostic of the disease.[2]
B. Acid alpha-mannosidase activity
Diagnosis is confirmed by measuring residual alpha-mannosidase activity in leukocytes or other nucleated cells via a fluorometric assay.[2] This is the most reliable diagnostic method, along with genetic testing.
C. Genetic testing
Identification of disease-causing mutations is achieved using DNA from peripheral blood cells, by polymerase chain reaction (PCR) amplification of all 24 MAN2B1 exons, followed by DNA sequencing.[2]
Treatment
There is no cure for congenital alpha-mannosidosis, and in general, the approach to management is proactive, with the aim of preventing emerging complications. After a full physical examination, physicians should focus on the known complications of alpha-mannosidosis, such as hydrocephalus, otitis media, hearing loss, dental caries, joint symptoms, kyphoscoliosis, and mental state.[2] Treatment is often limited to reducing or controlling the symptoms of the condition by, for example, medications to control seizures, hearing aids to ameliorate hearing loss, and routine physical therapy to assist with muscular pain and weakness.[2] In some cases, a wheelchair may be appropriate if muscle or spinal impairments immobilize the individual affected.
Hematopoietic stem cell transplantation (HSCT) can be a treatment option for some patients, however the risk-benefit profile is more favourable in younger patients, therefore ensuring an early diagnosis is critical for this to be a viable option.[2] The rationale is that enzyme-producing donor cells repopulate the host tissue and transfer enzyme to nearby enzyme-deficient host cells.[2] Despite early reports to the contrary,[9][10][2] The possible benefits of HSCT must be weighed against the overall risk of procedure related morbidity and mortality. The benefits are greater in younger patients before complications have developed, and also transplant related complications are more frequent and severe in older patients.
Enzyme replacement therapy (ERT) is a therapeutic alternative in a number of lysosomal storage diseases.[2][7] The overall principle of ERT is that a recombinantly produced version of the deficient enzyme is introduced into the blood stream, from where it is internalised by the cells and reaches the lysosomes by mannose-6-phosphate receptor mediated uptake, thus replacing the missing endogenous enzyme.[7] An ERT is approved for use in the European Union.[11]
Prognosis
The long-term forecast for the condition is poor.[2] There is generally a slow progression of neuromuscular and bone changes over decades. Behavioural problems, or psychiatric disorders may also be present.[2][7] The life expectancy in alpha-mannosidosis is highly variable. Individuals with early onset severe disease often do not survive beyond childhood, whereas those with milder disorders may survive well into adult life.
Independent living will be difficult, and patients with alpha-mannosidosis may become socially isolated, and during the late stages of the disease, they may become wheelchair bound, as they can no longer walk unaided.[2] This is likely to have a negative impact upon the quality of life of caregivers and family members.[2][7]
Epidemiology
The worldwide incidence of alpha-mannosidosis is not known exactly. However, a number of reports from different countries estimate that it occurs in approximately one in every million babies born worldwide.[8] Mannosidosis is found in all ethnic groups in Europe, America, Africa, and Asia.[2]
References
- Roces DP, Lüllmann-Rauch R, Peng J, et al. (2004). "Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study". Hum. Mol. Genet. 13 (18): 1979–88. doi:10.1093/hmg/ddh220. PMID 15269179.
- Malm D, Nilssen O (2008). "Alpha-mannosidosis". Orphanet J Rare Dis. 3 (1): 21. doi:10.1186/1750-1172-3-21. PMC 2515294 . PMID 18651971
- Gotoda Y, Wakamatsu N, Kawai H, Nishida Y, Matsumoto T (October 1998). "Missense and nonsense mutations in the lysosomal alpha-mannosidase gene (MANB) in severe and mild forms of alpha-mannosidosis". American Journal of Human Genetics. 63 (4): 1015–24. doi:10.1086/302048. PMC 1377481. PMID 9758606.
- Alpha-Mannosidosis Mutation Database. Tromsoe University. Available at: https://apex.jupiter.no/apex/f?p=101:1.
- Alpha Mannosidosis. National Organization for Rare Diseases (NORD) Factsheet 2015. https://rarediseases.org/rare-diseases/alpha-mannosidosis/
- Guide to understanding mannosidosis. Society for Mucopolysaccharide Diseases. http://www.mpssociety.org.uk/wp-content/uploads/2016/07/guide-alphamannosidosis-2013.pdf
- Borgwardt L, Lund AM, Dali CI (2014). Alpha-mannosidosis – a review of genetic, clinical findings and options for treatment. Pediatr. Endocrinol. Rev. 12 Suppl 1:185-91.
- Beck M Olsen KJ, Wraith JE et al. Natural history of alpha-mannosidosis: a longitudinal study. Orphanet J Rare Dis 2013;8:88.
- Will A, et al. (1987). "Bone marrow transplantation in the treatment of alpha-mannosidosis". Disease in Childhood. 62 (10): 1044–1049. doi:10.1136/adc.62.10.1044.
- Grewal SS, Shapiro EG, Krivit W, et al. (2004).Effective treatment of alpha-mannosidosis by allogeneic hematopoietic stem cell transplantation. J Pediatr, 144:569-573.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003922/human_med_002231.jsp&mid=WC0b01ac058001d124
Further reading
External links
| Classification | |
|---|---|
| External resources |