Amyloid plaques
Amyloid plaques (also known as neuritic plaques, Aβ plaques or senile plaques) are extracellular deposits of the amyloid beta (Aβ) protein mainly in the grey matter of the brain.[1][2][3][4] Degenerative neuronal elements and an abundance of microglia and astrocytes can be associated with amyloid plaques. Some plaques occur in the brain as a result of senescence (aging), but large numbers of plaques and neurofibrillary tangles are characteristic features of Alzheimer's disease.[5] Abnormal neurites in amyloid plaques are tortuous, often swollen axons and dendrites. The neurites contain a variety of organelles and cellular debris, and many of them include characteristic paired helical filaments, the ultrastructural component of neurofibrillary tangles.[3] The plaques are highly variable in shape and size; in tissue sections immunostained for Aβ, they comprise a log-normal size distribution curve with an average plaque area of 400-450 square micrometers (µm²). The smallest plaques (less than 200 µm²), which often consist of diffuse deposits of Aβ,[4] are particularly numerous.[6] The apparent size of plaques is influenced by the type of stain used to detect them, and by the plane through which they are sectioned for analysis under the microscope.[4] Plaques form when Aβ misfolds and aggregates into oligomers and longer polymers, the latter of which are characteristic of amyloid. Misfolded and aggregated Aβ is thought to be neurotoxic, especially in its oligomeric state.[7]
History
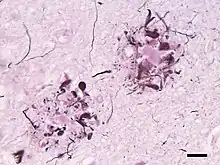
In 1892 Paul Blocq and Gheorghe Marinescu first described the presence of plaques in grey matter.[8][9] They referred to the plaques as 'nodules of neuroglial sclerosis'. In 1898, Emil Redlich reported plaques in three patients, two of whom had clinically verified dementia.[10] Redlich used the term 'miliary sclerosis' to describe plaques because he thought they resembled millet seeds, and he was the first to refer to the lesions as 'plaques'.[4] In the early 20th century, Oskar Fischer noted their similarity to actinomyces 'Drusen' (geode-like lesions), leading him to call the degenerative process 'drusige Nekrose'.[11] Alois Alzheimer is often credited with first linking plaques to dementia in a 1906 presentation (published in 1907),[12] but this short report focused mainly on neurofibrillary tangles, and plaques were only briefly mentioned.[4] Alzheimer's first substantive description of plaques appeared in 1911.[11] In contrast, Oskar Fischer published a series of comprehensive investigations of plaques and dementia in 1907, 1910 and 1912.[11] By 1911 Max Bielschowsky proposed the amyloid-nature of plaque deposits. This was later confirmed by Paul Divry, who showed that plaques that are stained with the dye Congo Red show the optical property of birefringence,[13] which is characteristic of amyloids in general.[14] In 1911, Teofil Simchowicz introduced the term 'senile plaques' to denote their frequent presence in the brains of older individuals.[15][16][17] In 1968, a quantitative analysis by Gary Blessed, Bernard Tomlinson and Martin Roth confirmed the association of senile plaques with dementia.[18] Henryk Wisniewski and Robert Terry coined the term 'neuritic plaques' in 1973 to designate plaques that include abnormal neuronal processes (neurites).[19] An important advance in 1984 and 1985 was the identification of Aβ as the protein that forms the cores of plaques.[20][21][22] This discovery led to the generation of new tools to study plaques, particularly antibodies to Aβ, and presented a molecular target for the development of potential therapies for Alzheimer's disease.[4] Knowledge of the amino acid sequence of Aβ also enabled scientists to discover genetic mutations that cause autosomal dominant Alzheimer's disease, all of which increase the likelihood that Aβ will aggregate in the brain.[23][24][25]
The generation of amyloid beta
Amyloid beta (Aβ) is a small protein, most often 40 or 42 amino acids in length, that is released from a longer parent protein called the Aβ-precursor protein (APP).[26] APP is produced by many types of cell in the body, but it is especially abundant in neurons. It is a single-pass transmembrane protein, that is, it passes once through cellular membranes.[27] The Aβ segment of APP is partly within the membrane and partly outside of the membrane. To liberate Aβ, APP is sequentially cleaved by two enzymes: first, by beta secretase (or β-amyloid cleaving enzyme (BACE) outside the membrane, and second, by gamma secretase (γ-secretase), an enzyme complex within the membrane.[27] The sequential actions of these secretases results in Aβ protein fragments that are released into the extracellular space[28][27] The discharge of Aβ is increased by the activity of synapses.[24] In addition to Aβ peptides that are 40 or 42 amino acids long, several less abundant Aβ fragments also are generated.[29][30] Aβ can be chemically modified in various ways, and the length of the protein and chemical modifications can influence both its tendency to aggregate and its toxicity.[4]
Identification
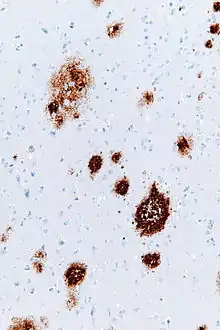
Amyloid plaques are visible with the light microscope using a variety of staining techniques, including silver stains, Congo red, Thioflavin, cresyl violet, PAS-reaction, and luminescent conjugated oligothiophenes (LCOs).[31][4][32] These methods often stain different components of the plaques, and they vary in their sensitivity[4][33] Plaques may also be visualized immunohistochemically with antibodies directed against Aβ or other components of the lesions. Immunohistochemical stains are especially useful because they are both sensitive and specific for antigens that are associated with plaques.
Composition
The Aβ deposits that comprise amyloid plaques are variable in size and appearance.[3][4] Under the light microscope, they range from small, wispy accumulations that are a few microns in diameter to much larger dense or diffuse masses. So-called 'classical plaques' consist of a compact Aβ-amyloid core that is surrounded by a corona of somewhat less densely packed Aβ.[4] Classical plaques also include abnormal, swollen neuronal processes (neurites) deriving from many different types of neurons, along with activated astrocytes and microglia.[3][4] Abnormal neurites and activated glial cells are not typical of most diffuse plaques, and it has been suggested that diffuse deposits are an early stage in the development of plaques.[34]
Anatomical distribution
Dietmar Thal and his colleagues have proposed a sequence of stages of plaque formation in the brains of Alzheimer patients[35][36] In Phase 1, plaques appear in the neocortex; in Phase 2, they appear in the allocortex, hippocampal formation and amygdala; in Phase 3, the basal ganglia and diencephalon are affected; in Phase 4, plaques appear in the midbrain and medulla oblongata; and in Phase 5, they appear in the pons and cerebellum. Thus, in end-stage Alzheimer's disease, plaques can be found in most parts of the brain. They are uncommon in the spinal cord.[4]
Formation and spread
The normal function of Aβ is not certain, but plaques arise when the protein misfolds and begins to accumulate in the brain by a process of molecular templating ('seeding').[37] Mathias Jucker and Lary Walker have likened this process to the formation and spread of prions in diseases known as spongiform encephalopathies or prion diseases.[37][38] According to the prion paradigm, certain proteins misfold into shapes that are rich in beta-sheet secondary structure. In this state, they cause other proteins of the same type to adopt the same abnormal beta-sheet-rich structure.[39] The misfolded proteins stick to one another, eventually stacking together to form protofibrils that twist together to make the amyloid fibrils that are typical of mature plaques.[40]
Involvement in disease
Abundant Aβ plaques, along with neurofibrillary tangles consisting of aggregated tau protein, are the two lesions that are required for the neuropathological diagnosis of Alzheimer's disease.[24][41] Although the number of neurofibrillary tangles correlates more strongly with the degree of dementia than does the number of plaques, genetic and pathologic findings indicate that Aβ plays a central role in the risk, onset, and progression of Alzheimer's disease.[23] Of particular importance is the longer (42 amino acids) species of Aβ known as Aβ42. Elevated levels of Aβ, as well as an increase in the ratio of Aβ42 to the 40-amino acid form (Aβ40), are important early events in the pathogenesis of Alzheimer's disease.[42]
Until recently, the diagnosis of Alzheimer's disease required a microscopic analysis of plaques and tangles in brain tissue, usually at autopsy.[43] However, Aβ plaques (along with cerebral Aβ-amyloid angiopathy) can now be detected in the brains of living subjects. This is done by preparing radiolabeled agents that bind selectively to Aβ deposits in the brain after being infused into the bloodstream.[44] The ligands cross the blood-brain barrier and attach to aggregated Aβ, and their retention in the brain is assessed by positron emission tomography (PET). In addition, the presence of plaques and tangles can be estimated by measuring the amounts of the Aβ and tau proteins in the cerebrospinal fluid.[45][46]
Occurrence
The probability of having plaques in the brain increases with advancing age.[47] From the age of 60 years (10%) to the age of 80 years (60%), the proportion of people with senile plaques increases linearly. Women are slightly more likely to have plaques than are men.[48][47] Both plaques and Alzheimer's disease also are more common in aging persons with trisomy-21 (Down syndrome).[1][49] This is thought to result from the excess production of Aβ because the APP gene is on chromosome 21, which exists as three copies in Down syndrome.[49]
Amyloid plaques naturally occur in the aging brains of nonhuman species ranging from birds to great apes.[4] In nonhuman primates, which are the closest biological relatives of humans, plaques have been found in all species examined thus far.[50] Neurofibrillary tangles are rare, however, and no nonhuman species has been shown to have dementia along with the complete neuropathology of Alzheimer's disease.[51]
Research
Research has been directed toward understanding the biochemical, cytological, and inflammatory characteristics of plaques, determining how plaques arise and proliferate in the brain, identifying genetic and environmental risk factors, discovering methods to detect them in the living brain, and developing therapeutic strategies for preventing or removing them.[4] Research on the formation and proliferation of amyloid plaques has been accelerated by the development of genetically modified mouse models.[52][53] Despite some limitations, these models have also contributed to the discovery of new therapeutic strategies. For example, a growing variety of treatments that reduce Aβ levels and the number of plaques in the brain have been identified with the help of transgenic rodent models. These strategies include immunotherapeutic approaches and inhibitors of the secretases that release Aβ from APP.[24] Such treatments are now being clinically evaluated for the treatment of Alzheimer's disease.[42][24] The findings so far indicate that the removal of plaques in patients with dementia is of little benefit, possibly because the brain is severely damaged by the time the signs and symptoms of Alzheimer's disease first appear.[24][23] For this reason, many researchers believe that earlier inhibition of Aβ aggregation and plaque formation is needed to slow or prevent tauopathy and the dementia of Alzheimer's disease. Other research is directed toward understanding the inflammation that is often associated with plaques[54] or identifying environmental, physiological and genetic risk factors for plaque formation and Alzheimer's disease.[55][56]
See also
References
- Cras P; Kawai M; Lowery D; Gonzalez-DeWhitt P; Greenberg B; Perry G (September 1991). "Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein". Proceedings of the National Academy of Sciences of the United States of America. 88 (17): 7552–6. Bibcode:1991PNAS...88.7552C. doi:10.1073/pnas.88.17.7552. PMC 52339. PMID 1652752.
- Purves, Dale; Augustine, George J.; Fitzpatrick, David; Hall, William C.; LaManita, Anthony-Samuel; White, Leonard E.; Mooney, Richard D.; Platt, Michael L. (2012). Neuroscience (5th ed.). Sunderland, MA: Sinauer Associates. p. 713. ISBN 978-0-87893-695-3.
- Dickson DW (1997). "The pathogenesis of senile plaques". J Neuropathol Exp Neurol. 56 (4): 321–339. doi:10.1097/00005072-199704000-00001. PMID 9100663.
- Walker LC (2020). "Aβ plaques". Free Neuropathology. 1 (31): 31. doi:10.17879/freeneuropathology-2020-3025. PMC 7745791. PMID 33345256.
- Ballard, C; Gauthier, S; Corbett, A; Brayne, C; Aarsland, D; Jones, E (19 March 2011). "Alzheimer's disease". Lancet. 377 (9770): 1019–31. doi:10.1016/S0140-6736(10)61349-9. PMID 21371747. S2CID 20893019.
- Hyman BT; West HL; Rebeck GW; Buldyrev SV; Mantegna RN; Ukleja M; Havlin S; Stanley HE (1995). "Quantitative analysis of senile plaques in Alzheimer disease: observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome)". Proceedings of the National Academy of Sciences of the United States of America. 92 (8): 3586–3590. Bibcode:1995PNAS...92.3586H. doi:10.1073/pnas.92.8.3586. PMC 42212. PMID 7724603.
- Haass C; Selkoe DJ (2007). "Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide". Nat Rev Mol Cell Biol. 8 (2): 101–112. doi:10.1038/nrm2101. PMID 17245412. S2CID 32991755.
- Blocq, Paul; Marinesco, Georges (1892). Sur les lesions et la pathogenie de l'epilepsie dite essentielle. pp. 445–6. OCLC 492619936.
- Buda O; Arsene D; Ceausu M; Dermengiu D; Curca GC (January 2009). "Georges Marinesco and the early research in neuropathology". Neurology. 72 (1): 88–91. doi:10.1212/01.wnl.0000338626.93425.74. PMID 19122036. S2CID 45428057.
- Redlich E (1898). "Ueber miliare Sklerose der Hirnrinde bei seniler Atrophie". Jahrbücher für Psychiatrie und Neurologie. 17: 208–216.
- Goedert M (2009). "Oskar Fischer and the study of dementia". Brain. 132 (4): 1102–1111. doi:10.1093/brain/awn256. PMC 2668940. PMID 18952676.
- Alzheimer, A (1907). "Uber einen eigenartige Erkranung der Hirnrinde". Allgemeine Zeitschrift für Psychiatrie und Psychisch-Gerichtlich Medizin. 64: 146–8.
- Divry P (1927). "Etude histo-chimique des plaques séniles". Journal Belge de Neurologie et de Psychiatrie. 9: 643–657.
- Buxbaum JN; Linke RP (2012). "A molecular history of the amyloidoses". Journal of Molecular Biology. 421 (2–3): 142–159. doi:10.1016/j.jmb.2012.01.024. PMID 22321796.
- Simchowicz T.: Histologische Studien über die senile Demenz. in: Nissl F., Alzheimer A. (Hrsg.): Histologische und histopathologische Arbeiten über die Grosshirnrinde mit besonderer Berücksichtigung der pathologischen Anatomie der Geisteskrankheiten. Jena: G. Fischer, 1911, p. 267–444.
- Ohry A; Buda O (2015). "Teofil Simchowicz (1879-1957): the scientist who coined senile plaques in neuropathology". Romanian Journal of Morphology and Embryology. 56 (4): 1545–1548. PMID 26743308.
- Grzybowski A; Pieta A; Pugaczewska M (2017). "Teofil Simchowicz (1879-1957)". Journal of Neurology. 264 (8): 1831–1832. doi:10.1007/s00415-017-8460-9. PMC 5533842. PMID 28315959.
- Blessed G; Tomlinson BE; Roth M (1968). "The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects". British Journal of Psychiatry. 114 (512): 797–811. doi:10.1192/bjp.114.512.797. PMID 5662937.
- Wisniewski, Henryk M.; Terry, Robert D. (1973). "Chapter 1: Reexamination of the pathogenesis of the senile plaque". In Zimmerman, H.M. (ed.). Progress in Neuropathology, Volume 2. Grune and Stratton. pp. 1–26. ISBN 978-0-808-90775-6.
- Glenner GG; Wong CW (1984). "Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein". Biochemical and Biophysical Research Communications. 120 (3): 885–890. doi:10.1016/s0006-291x(84)80190-4. PMID 6375662.
- Glenner GG; Wong CW (1984). "Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein". Biochemical and Biophysical Research Communications. 122 (3): 1131–1135. doi:10.1016/0006-291x(84)91209-9. PMID 6236805.
- Masters CL; Simms G; Weinman NA; Multhaup G; McDonald BL; Beyreuther K (1985). "Amyloid plaque core protein in Alzheimer disease and Down syndrome". Proceedings of the National Academy of Sciences USA. 82 (12): 4245–4249. Bibcode:1985PNAS...82.4245M. doi:10.1073/pnas.82.12.4245. PMC 397973. PMID 3159021.
- Walsh DM; Selkoe DJ (2020). "Amyloid beta-protein and beyond: the path forward in Alzheimer's disease". Current Opinion in Neurobiology. 61: 116–124. doi:10.1016/j.conb.2020.02.003. PMID 32197217. S2CID 214600892.
- Long JM; Holtzman DM (2019). "Alzheimer Disease: An Update on Pathobiology and Treatment Strategies". Cell. 179 (2): 312–339. doi:10.1016/j.cell.2019.09.001. PMC 6778042. PMID 31564456.
- Walker LC (2015). "Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases". Annual Review of Genetics. 50: 329–346. doi:10.1146/annurev-genet-120215-034943. PMC 6690197. PMID 27893962.
- Selkoe, DJ (1999). "Chapter 19: Biology of β-amyloid precursor protein and the mechanism of Alzheimer disease". In Terry, RD; Katzman, R; Bick, KL; Sisodia, SS (eds.). Alzheimer Disease. Lippincott Williams and Wilkins. pp. 293–310. ISBN 0-7817-1503-2.
- Haass C; Kaether C; Thinakaran G; Sisodia S (2012). "Trafficking and proteolytic processing of APP". Cold Spring Harbor Perspectives in Medicine. 2 (5:a006270): a006270. doi:10.1101/cshperspect.a006270. PMC 3331683. PMID 22553493.
- Suh YH; Checler F (September 2002). "Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer's disease". Pharmacological Reviews. 54 (3): 469–525. doi:10.1124/pr.54.3.469. PMID 12223532. S2CID 86686003.
- Dunys J; Valverde A; Checler F (2018). "Are N- and C-terminally truncated Aβ species key pathological triggers in Alzheimer's disease?". Journal of Biological Chemistry. 293 (40): 15419–15428. doi:10.1074/jbc.R118.003999. PMC 6177599. PMID 30143530.
- Kummer MP; Heneka MT (2014). "Truncated and modified amyloid-beta species". Alzheimers Research and Therapy. 6 (3): 28. doi:10.1186/alzrt258. PMC 4055046. PMID 25031638.
- Lamy C, Duyckaerts C, Delaere P, et al. (1989). "Comparison of seven staining methods for senile plaques and neurofibrillary tangles in a prospective series of 15 elderly patients". Neuropathology and Applied Neurobiology. 15 (6): 563–78. doi:10.1111/j.1365-2990.1989.tb01255.x. PMID 2482455. S2CID 25220224.
- Klingstedt T; Nilsson KPR (2012). "Luminescent conjugated poly- and oligo-thiophenes: optical ligands for spectral assignment of a plethora of protein aggregates". Biochemical Society Transactions. 40 (4): 704–710. doi:10.1042/BST20120009. PMID 22817720.
- Mavrogiorgou P; Gertz HJ; Ferszt R; Wolf R; Bär KJ; Juckel G (December 2011). "Are routine methods good enough to stain senile plaques and neurofibrillary tangles in different brain regions of demented patients?" (PDF). Psychiatria Danubina. 23 (4): 334–9. PMID 22075733.
- Braak H; Thal DR; Ghebremedhin E; Del Tredici K (2011). "Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years". Journal of Neuropathology and Experimental Neurology. 70 (11): 960–969. doi:10.1097/NEN.0b013e318232a379. PMID 22002422.
- Thal DR; Rüb O; Orantes M; Braak H (2002). "Phases of Abeta-deposition in the human brain and its relevance for the development of AD". Neurology. 58 (12): 1791–1800. doi:10.1212/wnl.58.12.1791. PMID 12084879. S2CID 41133337.
- Thal DR; Walter J; Saido TC; Fändrich M (2015). "Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer's disease". Acta Neuropathologica. 129 (2): 167–182. doi:10.1007/s00401-014-1375-y. PMID 25534025. S2CID 19701015.
- Jucker, M; Walker, LC (2013). "Self-propagation of pathogenic protein aggregates in neurodegenerative diseases". Nature. 501 (7465): 45–51. Bibcode:2013Natur.501...45J. doi:10.1038/nature12481. PMC 3963807. PMID 24005412.
- Walker LC; Jucker M (2015). "Neurodegenerative diseases: Expanding the prion concept". Annual Review of Neuroscience. 38: 87–103. doi:10.1146/annurev-neuro-071714-033828. PMC 4803040. PMID 25840008.
- Prusiner SB (1998). "Prions". Proceedings of the National Academy of Sciences USA. 95 (23): 13363–13383. Bibcode:1998PNAS...9513363P. doi:10.1073/pnas.95.23.13363. PMC 33918. PMID 9811807.
- Eisenberg D; Jucker M (2015). "The amyloid state of proteins in human diseases". Cell. 148 (6): 1188–1203. doi:10.1016/j.cell.2012.02.022. PMC 3353745. PMID 22424229.
- Nelson PT, Alafuzoff I, Bigio EH, et al. (2012). "Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature". Journal of Neuropathology and Experimental Neurology. 71 (5): 362–381. doi:10.1097/NEN.0b013e31825018f7. PMC 3560290. PMID 22487856.
- Findeis MA (November 2007). "The role of amyloid beta peptide 42 in Alzheimer's disease". Pharmacology & Therapeutics. 116 (2): 266–86. doi:10.1016/j.pharmthera.2007.06.006. PMID 17716740.
- Thal DR, Ronisz A, Tousseyn T, et al. (2019). "Different aspects of Alzheimer's disease-related amyloid β-peptide pathology and their relationship to amyloid positron emission tomography imaging and dementia". Acta Neuropathologica Communications. 7 (1): 178. doi:10.1186/s40478-019-0837-9. PMC 6854805. PMID 31727169.
- Mathis CA; Lopresti BJ; Ikonomovic MD; Klunk WE (2017). "Small-molecule PET tracers for imaging proteinopathies". Seminars in Nuclear Medicine. 47 (5): 553–575. doi:10.1053/j.semnuclmed.2017.06.003. PMC 5657567. PMID 28826526.
- Ritchie C; Smailagic N; Noel-Storr AH; Ukoumunne O; Ladds EC; Martin S (2017). "CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer's disease dementia and other dementias in people with mild cognitive impairment (MCI)". Cochrane Database of Systematic Reviews. 3 (3): CD010803. doi:10.1002/14651858.CD010803.pub2. PMC 6464349. PMID 28328043.
- Hansson O; Lehmann S; Otto M; Zetterberg H; Lewczuk P (2019). "Advantages and disadvantages of the use of the CSF Amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer's Disease". Alzheimer's Research and Therapy. 11 (1): 34. doi:10.1186/s13195-019-0485-0. PMC 6477717. PMID 31010420.
- Stam FC; Wigboldus JM; Smeulders AW (1986). "Age incidence of senile brain amyloidosis" (PDF). Pathology - Research and Practice. 181 (5): 558–562. doi:10.1016/S0344-0338(86)80149-2. PMID 3786248.
- Franke, M (1976). "Statistische Untersuchungen über die senilen Drusen im menschlichen Gehirn / Thesen". Berlin, Germany: Neuropathologische Abteilung. Archived from the original on 2011-07-19.
- Head E; Powell D; Gold BT; Schmitt FA (2012). "Alzheimer's Disease in Down Syndrome". European Journal of Neurodegenerative Disease. 1 (3): 353–364. PMC 4184282. PMID 25285303.
- Heuer E; Rosen RF; Cintron A; Walker LC (2012). "Nonhuman primate models of Alzheimer-like cerebral proteopathy". Current Pharmaceutical Design. 18 (8): 1159–1169. doi:10.2174/138161212799315885. PMC 3381739. PMID 22288403.
- Walker LC; Jucker M (2017). "The exceptional vulnerability of humans to Alzheimer's disease". Trends in Molecular Medicine. 23 (6): 534–545. doi:10.1016/j.molmed.2017.04.001. PMC 5521004. PMID 28483344.
- Jucker M (2010). "The benefits and limitations of animal models for translational research in neurodegenerative diseases". Nature Medicine. 16 (11): 1210–1214. doi:10.1038/nm.2224. PMID 21052075. S2CID 30167302.
- Myers A; McGonigle P (2010). "Overview of transgenic mouse models for Alzheimer's disease". Current Protocols in Neuroscience. 89 (1:e81): 1210–1214. doi:10.1002/cpns.81. PMID 31532917. S2CID 202024310.
- Heppner FL; Ransohoff RM; Becher B (2015). "Immune attack: the role of inflammation in Alzheimer disease". Nature Reviews Neuroscience. 16 (6): 358–372. doi:10.1038/nrn3880. PMID 25991443. S2CID 6116253.
- De Strooper B; Karran E (2016). "The Cellular Phase of Alzheimer's Disease". Cell. 164 (4): 603–615. doi:10.1016/j.cell.2015.12.056. PMID 26871627.
- Killin LOJ; Starr JM; Shiue IJ; Russ TC (2016). "Environmental risk factors for dementia: a systematic review". BMC Geriatrics. 16 (1): 175. doi:10.1186/s12877-016-0342-y. PMC 5059894. PMID 27729011.
Further reading
- Jellinger KA. Neurodegenerative Erkrankungen (ZNS) - Eine aktuelle Übersicht. Journal für Neurologie, Neurochirurgie und Psychiatrie. 2005;6(1):9-18.
- Cruz L, Urbanc B, Buldyrev SV, et al. (July 1997). "Aggregation and disaggregation of senile plaques in Alzheimer disease". Proceedings of the National Academy of Sciences of the United States of America. 94 (14): 7612–6. Bibcode:1997PNAS...94.7612C. doi:10.1073/pnas.94.14.7612. PMC 23870. PMID 9207140.
