Bayesian tool for methylation analysis
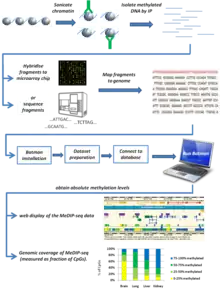
Bayesian tool for methylation analysis, also known as BATMAN, is a statistical tool for analysing methylated DNA immunoprecipitation (MeDIP) profiles. It can be applied to large datasets generated using either oligonucleotide arrays (MeDIP-chip) or next-generation sequencing (MeDIP-seq), providing a quantitative estimation of absolute methylation state in a region of interest.[1]
Theory
MeDIP (methylated DNA immunoprecipitation) is an experimental technique used to assess DNA methylation levels by using an antibody to isolate methylated DNA sequences. The isolated fragments of DNA are either hybridized to a microarray chip (MeDIP-chip) or sequenced by next-generation sequencing (MeDIP-seq). While this tells you what areas of the genome are methylated, it does not give absolute methylation levels. Imagine two different genomic regions, A and B. Region A has six CpGs (DNA methylation in mammalian somatic cells generally occurs at CpG dinucleotides[2]), three of which are methylated. Region B has three CpGs, all of which are methylated. As the antibody simply recognizes methylated DNA, it will bind both these regions equally and subsequent steps will therefore show equal signals for these two regions. This does not give the full picture of methylation in these two regions (in region A only half the CpGs are methylated, whereas in region B all the CpGs are methylated). Therefore, to get the full picture of methylation for a given region you have to normalize the signal you get from the MeDIP experiment to the number of CpGs in the region, and this is what the Batman algorithm does. Analysing the MeDIP signal of the above example would give Batman scores of 0.5 for region A (i.e. the region is 50% methylated) and 1 for region B (i.e. The region is 100% methylated). In this way Batman converts the signals from MeDIP experiments to absolute methylation levels.
Development of Batman
The core principle of the Batman algorithm is to model the effects of varying density of CpG dinucleotides, and the effect this has on MeDIP enrichment of DNA fragments. The basic assumptions of Batman:
- Almost all DNA methylation in mammals happens at CpG dinucleotides.
- Most CpG-poor regions are constitutively methylated while most CpG-rich regions (CpG islands) are constitutively unmethylated.[3]
- There are no fragment biases in MeDIP experiment (approximate range of DNA fragment sizes is 400–700 bp).
- The errors on the microarray are normally distributed with precision.
- Only methylated CpGs contribute to the observed signal.
- CpG methylation state is generally highly correlated over hundreds of bases,[4] so CpGs grouped together in 50- or 100-bp windows would have the same methylation state.
Basic parameters in Batman:
- Ccp: coupling factor between probe p and CpG dinucleotide c, is defined as the fraction of DNA molecules hybridizing to probe p that contain the CpG c.
- Ctot : total CpG influence parameter, is defined as the sum of coupling factors for any given probe, which provides a measure of local CpG density
- mc : the methylation status at position c, which represents the fraction of chromosomes in the sample on which it is methylated. mc is considered as a continuous variable since the majority samples used in MeDIP studies contain multiple cell-types.
Based on these assumptions, the signal from the MeDIP channel of the MeDIP-chip or MeDIP-seq experiment depends on the degree of enrichment of DNA fragments overlapping that probe, which in turn depends on the amount of antibody binding, and thus to the number of methylated CpGs on those fragments. In Batman model, the complete dataset from a MeDIP/chip experiment, A, can be represented by a statistical model in the form of the following probability distribution:

where (x|μ, σ2) is a Gaussian probability density function. Standard Bayesian techniques can be used to infer f(m|A), that is, the distribution of likely methylation states given one or more sets of MeDIP-chip/MeDIP-seq outputs. To solve this inference problem, Batman uses nested sampling (http://www.inference.phy.cam.ac.uk/bayesys/) to generate 100 independent samples from f(m|A) for each tiled region of the genome, then summarizes the most likely methylation state in 100-bp windows by fitting beta distributions to these samples. The modes of the most likely beta distributions were used as final methylation calls.

Limitations
It may be useful to take the following points into account when considering using Batman:
- Batman is not a piece of software; it is an algorithm performed using the command prompt. As such it is not especially user-friendly and is quite a computationally technical process.
- Because it is non-commercial, there is very little support when using Batman beyond what is in the manual.
- It is quite time-consuming (it can take several days to analyse one chromosome). (Note: In one government lab, running Batman on a set of 100 Agilent Human DNA Methylation Arrays (about 250,000 probes per array) took less than an hour to complete in Agilent's Genomic Workbench software. Our computer had a 2GHz processor, 24 GB RAM, 64-bit Windows 7.)
- Copy number variation (CNV) has to be accounted for. For example, the score for a region with a CNV value of 1.6 in a cancer (a loss of 0.4 compared to normal) would have to be multiplied by 1.25 (=2/1.6) to compensate for the loss.
- One of the basic assumptions of Batman is that all DNA methylation occurs at CpG dinucleotides. While this is generally the case for vertebrate somatic cells, there are situations where there is widespread non-CpG methylation, such as in plant cells and embryonic stem cells.[5][6]
References
- Down, T.A. et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nature Biotechnology 26, 779–85 (2008).
- Lister, R. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–22 (2009).
- Bird, A. DNA methylation patterns and epigenetic memory. Genes & Development 16, 6–21 (2002).
- Eckhardt, F. et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nature Genetics 38, 1378–85 (2006).
- Dodge, J.E., Ramsahoye, B.H., Wo, Z.G., Okano, M. & Li, E. De novo methylation of MMLV provirus in embryonic stem cells: CpG versus non-CpG methylation. Gene 289, 41–8 (2002)
- Vanyushin, B.F. DNA methylation in plants. Current Topics in Microbiology and Immunology 301, 67–122 (2006)
