CTNS (gene)
CTNS may also refer to the Center for Theology and the Natural Sciences.


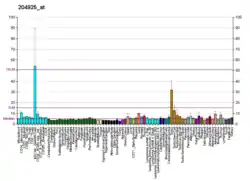
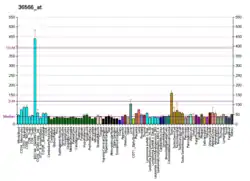
CTNS is the gene that encodes the protein cystinosin in humans. Cystinosin is a lysosomal seven-transmembrane protein that functions as an active transporter for the export of cystine molecules out of the lysosome.
Mutations in CTNS are responsible for cystinosis, an autosomal recessive lysosomal storage disease.[5]
Discovery
In 1995, the gene was localized to the short arm of chromosome 17.[6] An international collaborative effort finally succeeded in isolating CTNS by positional cloning in 1998.[5]
Gene
CTNS is located on the p arm of human chromosome 17, at position 13.2.[5] It spans base pairs 3,636,468 and 3,661,542, and comprises 12 exons.[5][7]
Tissue distribution
The gene is expressed in the lysosomes of all organs and tissues.[8] Cystinosin has also been found in melanosomes in melanocytes.[9]
Structure
Cystinosin is a seven-transmembrane domain receptor embedded in the lysosomal membrane, and is a member of the lysosomal cystine transporter family of transport proteins.[10] It comprises 367 amino acid residues, and has a molecular mass of 41738 Da.[10] Cystinosin has seven N-glycosylation sites in the N-terminus region, spanning a range of 128 amino acid residues.[11]
The receptor also has two sorting motifs; a GYDQL motif in the C-terminus region, and a YFPQA motif, known as the 'PQ loop,' on the fifth inter-transmembrane α-helix moiety.[12]
Mechanism
The protein obeys Michaelis-Menton kinetics and has an associated KM of 278 ± 49 μM.[11][13]
Function
Cystinosin functions as a symporter which actively transports protons and cystine, the oxidized cysteine dimer, out of the lysosome.[11] This is necessary to distribute cystine to the rest of the cell and allow the lysosome to continue to function.
Cystinosin has also been discovered in melanosomes and has been linked to the control and regulation of melanin.[9]
Clinical significance
Cystinosis
Mutations in CTNS can result in cystinosis. Cystinosis is a type of lysosomal transport disorder, a subset of lysosomal storage disorders.[14] Variation in the encoded cystinosin protein results in an inhibition or loss in its ability to transport cystine out of the lysosome. Cystine molecules accumulate and form crystals within the lysosome, impairing its function.[8]
Mutations
Cystinosis is presented in patients with a range of CTNS mutations; as of 2017, over 100 have been identified.[15][16] The most common mutation is a 57,257 base pair deletion commonly referred to as the 57 kb deletion. This was formally known as the 65 kb deletion; a misnomer originating from early incorrect estimates.[17][18] Other reported mutations include other deletions, missense mutations, and in-frame deletions and insertions.[19][20]
The type and extent of mutation determines the type and severity of cystinosis in the carrier.[21] This is a result of the degree of transport inhibition caused by the misfolding of cystinosin.[19] For example, mild cystinosis is typically associated with mutations that do not affect the amino acids in the transmembrane domains of cystinosin.[7] In contrast, infantile nephropathic cystinosis, the most severe form of the disease, is most commonly associated with a total loss of activity.[19]
Gene deletion resulting in the absence of either of the sorting motifs results in the delocalization of cystinosin to the cellular plasma membrane.[22][11]
Model systems
Human models for cystinosin are typically derived from cystinotic renal tubular cell lines.[23][24]
Non-human protein homologs for cystinosin include ERS1 in Saccharomyces cerevisiae (yeast cells) and the Caenorhabditis elegans protein, C41C4.7.[25] Murine ctns has also been used.[26]
References
- GRCh38: Ensembl release 89: ENSG00000040531 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000005949 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M, Bates GP, van't Hoff W, Antignac C (April 1998). "A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis". Nature Genetics. 18 (4): 319–24. doi:10.1038/ng0498-319. PMID 9537412. S2CID 10629789.
- McDowell GA, Gahl WA, Stephenson LA, Schneider JA, Weissenbach J, Polymeropoulos MH, Town MM, William, Hoff T, Farrall M, Mathew CG (June 1995). "Linkage of the gene for cystinosis to markers on the short arm of chromosome 17. The Cystinosis Collaborative Research Group". Nature Genetics. 10 (2): 246–8. doi:10.1038/ng0695-246. PMID 7663525. S2CID 22093385.
- Shotelersuk V, Larson D, Anikster Y, McDowell G, Lemons R, Bernardini I, Guo J, Thoene J, Gahl WA (November 1998). "CTNS mutations in an American-based population of cystinosis patients". American Journal of Human Genetics. 63 (5): 1352–62. doi:10.1086/302118. PMC 1377545. PMID 9792862.
- Nesterova G, Gahl WA (January 2013). "Cystinosis: the evolution of a treatable disease". Pediatric Nephrology. 28 (1): 51–9. doi:10.1007/s00467-012-2242-5. PMC 3505515. PMID 22903658.
- Chiaverini C, Sillard L, Flori E, Ito S, Briganti S, Wakamatsu K, Fontas E, Berard E, Cailliez M, Cochat P, Foulard M, Guest G, Niaudet P, Picardo M, Bernard FX, Antignac C, Ortonne JP, Ballotti R (September 2012). "Cystinosin is a melanosomal protein that regulates melanin synthesis". FASEB Journal. 26 (9): 3779–89. doi:10.1096/fj.11-201376. PMID 22649030. S2CID 11334825.
- "Transporter Classification Database". www.tcdb.org. 2017-10-13. Archived from the original on 2014-01-03. Retrieved 2017-10-13.
- Kalatzis V, Cherqui S, Antignac C, Gasnier B (November 2001). "Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter". The EMBO Journal. 20 (21): 5940–9. doi:10.1093/emboj/20.21.5940. PMC 125690. PMID 11689434.
- Andrzejewska Z, Névo N, Thomas L, Bailleux A, Chauvet V, Benmerah A, Antignac C (July 2015). "Lysosomal Targeting of Cystinosin Requires AP-3". Traffic. 16 (7): 712–26. doi:10.1111/tra.12277. PMID 25753619.
- Ruivo R, Bellenchi GC, Chen X, Zifarelli G, Sagné C, Debacker C, Pusch M, Supplisson S, Gasnier B (January 2012). "Mechanism of proton/substrate coupling in the heptahelical lysosomal transporter cystinosin". Proceedings of the National Academy of Sciences of the United States of America. 109 (5): E210-7. doi:10.1073/pnas.1115581109. PMC 3277178. PMID 22232659.
- Mancini GM, Havelaar AC, Verheijen FW (May 2000). "Lysosomal transport disorders". Journal of Inherited Metabolic Disease. 23 (3): 278–92. doi:10.1023/a:1005640214408. PMID 10863944. S2CID 19489712.
- Doneray H, Aldahmesh M, Yilmaz G, Cinici E, Orbak Z (June 2017). "Infantile Nephropathic Cystinosis: A Novel CTNS Mutation". The Eurasian Journal of Medicine. 49 (2): 148–151. doi:10.5152/eurasianjmed.2017.17039. PMC 5469843. PMID 28638260.
- Owen EP, Nandhlal J, Leisegang F, Van der Watt G, Nourse P, Gajjar P (April 2015). "Common mutation causes cystinosis in the majority of black South African patients". Pediatric Nephrology. 30 (4): 595–601. doi:10.1007/s00467-014-2980-7. PMID 25326109. S2CID 22240586.
- Touchman JW, Anikster Y, Dietrich NL, Maduro VV, McDowell G, Shotelersuk V, Bouffard GG, Beckstrom-Sternberg SM, Gahl WA, Green ED (February 2000). "The genomic region encompassing the nephropathic cystinosis gene (CTNS): complete sequencing of a 200-kb segment and discovery of a novel gene within the common cystinosis-causing deletion". Genome Research. 10 (2): 165–73. doi:10.1101/gr.10.2.165. PMC 310836. PMID 10673275.
- Anikster Y, Lucero C, Touchman JW, Huizing M, McDowell G, Shotelersuk V, Green ED, Gahl WA (February 1999). "Identification and detection of the common 65-kb deletion breakpoint in the nephropathic cystinosis gene (CTNS)". Molecular Genetics and Metabolism. 66 (2): 111–6. doi:10.1006/mgme.1998.2790. PMID 10068513.
- Kalatzis V, Nevo N, Cherqui S, Gasnier B, Antignac C (July 2004). "Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin". Human Molecular Genetics. 13 (13): 1361–71. doi:10.1093/hmg/ddh152. PMID 15128704.
- Tang S, Danda S, Zoleikhaeian M, Simon M, Huang T (August 2009). "An Indian boy with nephropathic cystinosis: a case report and molecular analysis of CTNS mutation". Genetic Testing and Molecular Biomarkers. 13 (4): 435–8. doi:10.1089/gtmb.2008.0156. PMID 19580442.
- Attard M, Jean G, Forestier L, Cherqui S, van't Hoff W, Broyer M, Antignac C, Town M (December 1999). "Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin". Human Molecular Genetics. 8 (13): 2507–14. doi:10.1093/hmg/8.13.2507. PMID 10556299.
- Cherqui S, Kalatzis V, Trugnan G, Antignac C (April 2001). "The targeting of cystinosin to the lysosomal membrane requires a tyrosine-based signal and a novel sorting motif". The Journal of Biological Chemistry. 276 (16): 13314–21. doi:10.1074/jbc.m010562200. PMID 11150305.
- Racusen LC, Wilson PD, Hartz PA, Fivush BA, Burrow CR (August 1995). "Renal proximal tubular epithelium from patients with nephropathic cystinosis: immortalized cell lines as in vitro model systems". Kidney International. 48 (2): 536–43. doi:10.1038/ki.1995.324. PMID 7564123.
- Taub ML, Springate JE, Cutuli F (April 2011). "Reduced phosphate transport in the renal proximal tubule cells in cystinosis is due to decreased expression of transporters rather than an energy defect". Biochemical and Biophysical Research Communications. 407 (2): 355–9. doi:10.1016/j.bbrc.2011.03.022. PMID 21392501.
- Gao XD, Wang J, Keppler-Ross S, Dean N (May 2005). "ERS1 encodes a functional homologue of the human lysosomal cystine transporter". The FEBS Journal. 272 (10): 2497–511. doi:10.1111/j.1742-4658.2005.04670.x. PMID 15885099. S2CID 13035448.
- Cherqui S, Sevin C, Hamard G, Kalatzis V, Sich M, Pequignot MO, Gogat K, Abitbol M, Broyer M, Gubler MC, Antignac C (November 2002). "Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis". Molecular and Cellular Biology. 22 (21): 7622–32. doi:10.1128/MCB.22.21.7622-7632.2002. PMC 135682. PMID 12370309.
Further reading
- Anikster Y, Shotelersuk V, Gahl WA (2000). "CTNS mutations in patients with cystinosis". Human Mutation. 14 (6): 454–8. doi:10.1002/(SICI)1098-1004(199912)14:6<454::AID-HUMU2>3.0.CO;2-H. PMID 10571941.
- Gahl WA, Thoene JG, Schneider JA (July 2002). "Cystinosis". The New England Journal of Medicine. 347 (2): 111–21. doi:10.1056/NEJMra020552. PMID 12110740.
- Kalatzis V, Antignac C (November 2002). "Cystinosis: from gene to disease". Nephrology, Dialysis, Transplantation. 17 (11): 1883–6. doi:10.1093/ndt/17.11.1883. PMID 12401840.
- Thoene J, Lemons R, Anikster Y, Mullet J, Paelicke K, Lucero C, Gahl W, Schneider J, Shu SG, Campbell HT (August 1999). "Mutations of CTNS causing intermediate cystinosis". Molecular Genetics and Metabolism. 67 (4): 283–93. doi:10.1006/mgme.1999.2876. PMID 10444339.
- McGowan-Jordan J, Stoddard K, Podolsky L, Orrbine E, McLaine P, Town M, Goodyer P, MacKenzie A, Heick H (September 1999). "Molecular analysis of cystinosis: probable Irish origin of the most common French Canadian mutation". European Journal of Human Genetics. 7 (6): 671–8. doi:10.1038/sj.ejhg.5200349. PMID 10482956.
- Anikster Y, Lucero C, Guo J, Huizing M, Shotelersuk V, Bernardini I, McDowell G, Iwata F, Kaiser-Kupfer MI, Jaffe R, Thoene J, Schneider JA, Gahl WA (January 2000). "Ocular nonnephropathic cystinosis: clinical, biochemical, and molecular correlations". Pediatric Research. 47 (1): 17–23. doi:10.1203/00006450-200001000-00007. PMID 10625078.
- Cherqui S, Kalatzis V, Forestier L, Poras I, Antignac C (2003). "Identification and characterisation of the murine homologue of the gene responsible for cystinosis, Ctns". BMC Genomics. 1: 2. doi:10.1186/1471-2164-1-2. PMC 29086. PMID 11121245.
- Phornphutkul C, Anikster Y, Huizing M, Braun P, Brodie C, Chou JY, Gahl WA (October 2001). "The promoter of a lysosomal membrane transporter gene, CTNS, binds Sp-1, shares sequences with the promoter of an adjacent gene, CARKL, and causes cystinosis if mutated in a critical region". American Journal of Human Genetics. 69 (4): 712–21. doi:10.1086/323484. PMC 1226058. PMID 11505338.
- Rupar CA, Matsell D, Surry S, Siu V (September 2001). "A G339R mutation in the CTNS gene is a common cause of nephropathic cystinosis in the south western Ontario Amish Mennonite population". Journal of Medical Genetics. 38 (9): 615–6. doi:10.1136/jmg.38.9.615. PMC 1734937. PMID 11565547.
- Kleta R, Anikster Y, Lucero C, Shotelersuk V, Huizing M, Bernardini I, Park M, Thoene J, Schneider J, Gahl WA (November 2001). "CTNS mutations in African American patients with cystinosis". Molecular Genetics and Metabolism. 74 (3): 332–7. doi:10.1006/mgme.2001.3218. PMID 11708862.
- Kiehntopf M, Schickel J, Gönne B, Koch HG, Superti-Furga A, Steinmann B, Deufel T, Harms E (September 2002). "Analysis of the CTNS gene in patients of German and Swiss origin with nephropathic cystinosis". Human Mutation. 20 (3): 237. doi:10.1002/humu.9063. PMID 12204010. S2CID 25080378.
External links
- GeneReviews/NCBI/NIH/UW entry on Cystinosis
- Human CTNS genome location and CTNS gene details page in the UCSC Genome Browser.
- Genetics Home Reference page on CTNS.
- Genetic Testing Registry.