Copper protein
Copper proteins are proteins that contain one or more copper ions as prosthetic groups. Copper proteins are found in all forms of air-breathing life. These proteins are usually associated with electron-transfer with or without the involvement of oxygen (O2). Some organisms even use copper proteins to carry oxygen instead of iron proteins. A prominent copper proteins in humans is in cytochrome c oxidase (cco). The enzyme cco mediates the controlled combustion that produces ATP.[1]
Classes
The metal centers in the copper proteins can be classified into several types:[2]
- Type I copper centres (T1Cu) are characterized by a single copper atom coordinated by two histidine residues and a cysteine residue in a trigonal planar structure, and a variable axial ligand. In class I T1Cu proteins (e.g. amicyanin, plastocyanin and pseudoazurin) the axial ligand is the sulfur of methionine, whereas aminoacids other than methionine (e.g. glutamine) give rise to class II T1Cu copper proteins. Azurins contain the third type of T1Cu centres: besides a methionine in one axial position, they contain a second axial ligand (a carbonyl group of a glycine residue). T1Cu-containing proteins are usually called "cupredoxins", and show similar three-dimensional structures, relatively high reduction potentials (> 250 mV), and strong absorption near 600 nm (due to S→Cu charge transfer), which usually gives rise to a blue colour. Cupredoxins are therefore often called "blue copper proteins". This may be misleading, since some T1Cu centres also absorb around 460 nm and are therefore green. When studied by EPR spectroscopy, T1Cu centres show small hyperfine splittings in the parallel region of the spectrum (compared to common copper coordination compounds).[3]
- Type II copper centres (T2Cu) exhibit a square planar coordination by N or N/O ligands. They exhibit an axial EPR spectrum with copper hyperfine splitting in the parallel region similar to that observed in regular copper coordination compounds. Since no sulfur ligation is present, the optical spectra of these centres lack distinctive features. T2Cu centres occur in enzymes, where they assist in oxidations or oxygenations.[4]
- Type III copper centres (T3Cu) consist of a pair of copper centres, each coordinated by three histidine residues. These proteins exhibit no EPR signal due to strong antiferromagnetic coupling (i.e. spin pairing) between the two S = 1/2 metal ions due to their covalent overlap with a bridging ligand. These centres are present in some oxidases and oxygen-transporting proteins (e.g. hemocyanin and tyrosinase).[5]
- Binuclear Copper A centres (CuA) are found in cytochrome c oxidase and nitrous-oxide reductase (EC 1.7.99.6). The two copper atoms are coordinated by two histidines, one methionine, a protein backbone carbonyl oxygen, and two bridging cysteine residues.[6]
- Copper B centres (CuB) are found in cytochrome c oxidase. The copper atom is coordinated by three histidines in trigonal pyramidal geometry.
- A tetranuclear Copper Z centre (CuZ) is found in nitrous-oxide reductase. The four copper atoms are coordinated by seven histidine residues and bridged by a sulfur atom.
Blue copper proteins
The blue copper proteins owe their name to their intense blue coloration (Cu(II)). The blue copper protein often called as “moonlighting protein”, which means a protein can perform more than one function. They serve as electron transfer agents, with the active site shuttling between Cu(I) and Cu(II). The Cu2+ in the oxidized state can accept one electron to form Cu1+ in the reduced protein. The geometry of the Cu center has a major impact on its redox properties. The Jahn-Teller distortion does not apply to the blue copper proteins because the copper site has low symmetry that does not support degeneracy in the d-orbital manifold. The absence of large reorganizational changes enhances the rate of their electron transfer. The active site of a type-I blue copper protein. Two 2-histidines, 1 methionine and 1 cysteine present in the coordination sphere. Example for Type-I blue copper protein are plastocyanine , azurin, and nitrite reductase. Thaemocyanin and tyrosinase .
Blue copper protein types structure
Blue Copper Proteins, a class of Type 1 copper proteins, are small proteins containing a cupredoxin fold and a single Type I copper ion coordinated by two histidine N-donors, a cysteine thiolate S-donor and a methionine thioether S-donor.[7] In the oxidized state, the Cu+2 ion will form either a trigonal bipyramidal or tetrahedral coordination.[7] The Type 1 copper proteins are identified as blue copper proteins due to the ligand to metal charge transfer an intense band at 600 nm that gives the characteristic of a deep blue colour present in the electron absorption spectrum.[8]
The protein structure of a Type 1 blue copper protein, amicyanin, is built off of polypeptide folds that are commonly found in blue copper proteins β sandwich structure.[9] The structure is very similar to plastocyanin and azurin as they also identify as Type 1 copper proteins.[9] They are also similar to one another due to the geometry of the copper site of each copper protein. The protein azurin has a trigonal bipyramidal geometry with elongated axial glycine and methoinione sulfur ligands. Plastocyanins have an additional methionine sulfur ligand on the axial position. The main difference of each copper protein is that each protein has different number and species of ligand coordinated to the copper center.
Electronic structure of the blue copper protein type I copper complexes
The strong bond between the copper ion and the cysteine sulfur allows for the non-bonded electron on the cysteine sulfur to be present on both the low/high spin state copper ion, dx2-dy2 orbital and the p-orbital of the cysteine sulfur.[8] Most copper (II) complexes will exhibit the Jahn-Teller effect when the complex forms a tetragonal distortion of an octahedral complex geometry.[10] With blue copper proteins, a distorted tetrahedral complex will be formed due to the strong equatorial cysteine ligand and the weak axial methionine ligand.[10] The two neutral histidine ligands are positioned by the protein ligand so the geometry is distorted tetrahedral. This will cause them not to be able to coordinate perfectly as tetrahedral or a square planar.
Spectral changes with temperature
Lowering the temperature may change the transitions. The intense absorbance at about 16000 cm−1 was characterized the absorptions feature of blue copper. There was a second lower energy feature band with moderate absorption intensity. Polarized signal-crystal absorption data on plasto-cyanin showed that both bands have the same polarization ratio that associated with Cu(II)-S(Cys) bond. This is explained that the normal cupric complex has high energy intense sigma and low energy weak π bonds. However, in the blue copper protein case have low energy intense sigma and high energy weak π bonds because CT intensity reflects overlap of the donor and acceptor orbitals in the CT process. This required that the 3d(x2-y2 ) orbital of the blue copper site be oriented such that its lobes bisect the Cu-S(Cys) bond giving dominant π overlap with sulfur directly. Finally, the nature of the ground state wave function of the blue copper protein is rich in electron absorption spectrum.
Inner and outer sphere metal coordination
The cysteine sulfur copper (II) ion bonds range from 2.6 to 3.2 Å.[11] With the reduced form, CuI, protein structures are still formed with elongated bonds by 0.1 Å or less. with the oxidized and reduced protein structures, they are superimposable. With amicyanin, there is an exception due to the histidine being ligated and it is not bound to copper iodide.[11] In azurin, the Cysteine112 thiolate accepts the hydrogen bonds from the amide backbone of Asparagine47, and Phenylalanine114, and Histidine46 donates a hydrogen bond to the carbonyl backbone of Asparagine10. The Cysteine84 thiolate of plastocyanin accepts a hydrogen bond from a amide backbone, Asparagine38, and Histidine37 interacts strongly with the carbonyl backbone of Alanine33 and more weakly with the carbonyl backbone of Leucine5, Glycine34, and the amide backbone of Phenylalanine35.[11]
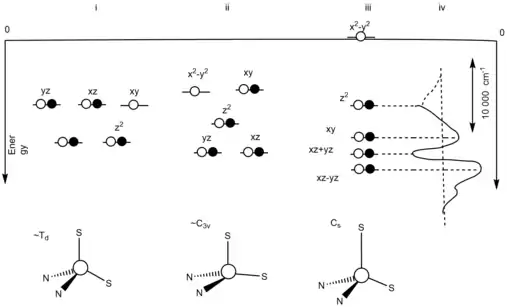
Blue copper protein ligand field effect
The orbital degeneracy is removed due to the asymmetric ligand field.[10] The asymmetric ligand field is influenced by the strong equatorial cysteine ligand and the weak axial methionine ligand. The reorganization of the oxidized, Cu+2, state, at the blue copper protein active site will be minimized due to the fact that at the oxidized, Cu+2, state, the Jahn-Teller effect will be ineffective.[10] In Figure 2, an energy level diagram is present to show the three different ideal geometries and its degenerate states.[10] (i) represents the energy level diagram of a tetrahedral geometric structure with a T2 degenerate ground state. This is due to the Jahn-Teller distortion from the oxidation. (ii) represents the energy level diagram of a C3v symmetric structure with a 2E degenerate ground state. This resulted due to the thioether bond being elongated in the reduction site of the blue copper protein. The unpaired electrons leads to the Jahn-Teller effect. (iii) represents the energy level diagram of the ground states not being on an equal level. This shows that there is no presence of the Jahn-Teller effect. This is due to the strong equatorial donor and weak axial donor interactions. (iv) represents the difference in distance between the dxy and dx2-y2.[10]
See also
References
- Lontie R, ed. (2018). Copper Proteins and Copper Enzymes. III. CRC Press. ISBN 9781315891798.
- Holm RH, Kennepohl P, Solomon EI (November 1996). "Structural and Functional Aspects of Metal Sites in Biology". Chemical Reviews. 96 (7): 2239–2314. doi:10.1021/cr9500390. PMID 11848828.
- Arcos-López, Trinidad; Schuth, Nils; Quintanar, Liliana (2020), "Chapter 3: The Type 1 Blue Copper Site: From Electron Transfer to Biological Function", in Sosa Torres, Martha E.; Kroneck, Peter M.H. (eds.), Transition Metals and Sulfur: A Strong Relationship for Life, Metal Ions in Life Sciences (Series editors Astrid Sigel, Eva Freisinger and Roland K.O. Sigel), 20, Berlin/Boston: de Gruyter, doi:10.1515/9783110589757-003
- Klinman JP (November 1996). "Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates". Chemical Reviews. 96 (7): 2541–2562. doi:10.1021/cr950047g. PMID 11848836..
- Lewis EA, Tolman WB (2004). "Reactivity of Dioxygen-Copper Systems". Chemical Reviews. 104 (2): 1047–1076. doi:10.1021/cr020633r. PMID 14871149.
- Solomon EI, Sundaram UM, Machonkin TE (November 1996). "Multicopper Oxidases and Oxygenases". Chemical Reviews. 96 (7): 2563–2606. doi:10.1021/cr950046o. PMID 11848837.
- Malmström BG (1994). "Rack-induced bonding in blue-copper proteins". EJB Reviews 1994. Berlin Heidelberg: Springer. pp. 157–164. doi:10.1007/978-3-642-79502-2_12. ISBN 978-3-540-58830-6.
- Bertini I (2007-07-01). "Biological inorganic chemistry: structure and reactivity". Choice Reviews Online. 44 (11): 44–6242–44-6242. doi:10.5860/CHOICE.44-6242. ISSN 0009-4978. S2CID 93183803.
- De Rienzo F, Gabdoulline RR, Menziani MC, Wade RC (August 2000). "Blue copper proteins: a comparative analysis of their molecular interaction properties". Protein Science. 9 (8): 1439–54. doi:10.1110/ps.9.8.1439. PMC 2144732. PMID 10975566.
- Solomon EI, Hadt RG (April 2011). "Recent advances in understanding blue copper proteins". Coordination Chemistry Reviews. 255 (7–8): 774–789. doi:10.1016/j.ccr.2010.12.008.
- Warren JJ, Lancaster KM, Richards JH, Gray HB (October 2012). "Inner- and outer-sphere metal coordination in blue copper proteins". Journal of Inorganic Biochemistry. 115: 119–26. doi:10.1016/j.jinorgbio.2012.05.002. PMC 3434318. PMID 22658756.