Crossover experiment (chemistry)
In chemistry, a crossover experiment is a method used to study the mechanism of a chemical reaction. In a crossover experiment, two similar but distinguishable reactants simultaneously undergo a reaction as part of the same reaction mixture. The products formed will either correspond directly to one of the two reactants (non-crossover products) or will include components of both reactants (crossover products). The aim of a crossover experiment is to determine whether or not a reaction process involves a stage where the components of each reactant have an opportunity to exchange with each other.
The results of crossover experiments are often straightforward to analyze, making them one of the most useful and most frequently applied methods of mechanistic study. In organic chemistry crossover experiments are most often used to distinguish between intramolecular and intermolecular reactions.[1][2][3] Inorganic and organometallic chemists rely heavily on crossover experiments, and in particular isotopic labeling experiments, for support or contradiction of proposed mechanisms.[4] When the mechanism being investigated is more complicated than an intra- or intermolecular substitution or rearrangement, crossover experiment design can itself become a challenging question.[5] A well-designed crossover experiment can lead to conclusions about a mechanism that would otherwise be impossible to make. Many mechanistic studies include both crossover experiments and measurements of rate and kinetic isotope effects.
Purpose
Crossover experiments allow for experimental study of a reaction mechanism. Mechanistic studies are of interest to theoretical and experimental chemists for a variety of reasons including prediction of stereochemical outcomes, optimization of reaction conditions for rate and selectivity, and design of improved catalysts for better turnover number, robustness, etc.[6][7] Since a mechanism cannot be directly observed or determined solely based on the reactants or products, mechanisms are challenging to study experimentally. Only a handful of experimental methods are capable of providing information about the mechanism of a reaction, including crossover experiments, studies of the kinetic isotope effect, and rate variations by substituent. The crossover experiment has the advantage of being conceptually straightforward and relatively easy to design, carry out, and interpret. In modern mechanistic studies, crossover experiments and KIE studies are commonly used in conjunction with computational methods.[8]
Theory
The concept underlying the crossover experiment is a basic one: provided that the labeling method chosen does not affect the way a reaction proceeds, a shift in the labeling as observed in the products can be attributed to the reaction mechanism. The most important limitation in crossover experiment design is therefore that the labeling not affect the reaction mechanism itself.[1]
It can be difficult to know whether or not the changes made to reactants for a crossover experiment will affect the mechanism by which the reaction proceeds. This is particularly true since the aim of the crossover experiment is to provide insight into the mechanism that would allow these types of predictions. There is always the possibility that a label will alter the course of the reaction.[1]
In practice, crossover experiments aim to use the least change possible between the usual conditions of the reaction being studied and the conditions of the crossover experiment. This principle favors isotopic labeling, since changing the isotope of one atom in a molecule is the smallest change that can be both easily enacted and traced in the reaction. If the isotope is placed in the molecule at a position directly involved in the mechanism of the reaction, a kinetic isotope effect is expected. This can be used to study aspects of the mechanism independently or alongside a crossover experiment.[1][2][8] The kinetic isotope effect is a change in the rate of reaction based on the change in isotope, not a change in the mechanism of the reaction itself, so isotopic labeling generally satisfies the requirements for a valid crossover experiment. In crossover experiments that do not use isotopic labeling, addition or subtraction of a methyl substituent at a position not involved in any proposed mechanism for the reaction is typically expected to give a valid crossover experiment.[1]
Design
In designing a crossover experiment the first task is to propose possible mechanisms for the reaction being studied. Based on these possible mechanisms, the goal is to determine either a traditional crossover experiment or an isotope scrambling experiment that will enable the researcher to distinguish between the two or more possible mechanisms. Often many methods of mechanistic study will have to be employed to support or discount all of the mechanisms proposed. However, in some cases a crossover experiment alone will be able to distinguish between the main possibilities, for example in the case of intramolecular vs. intermolecular organic reaction mechanisms.
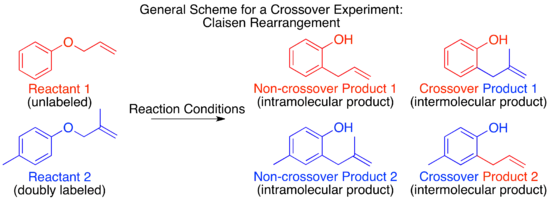
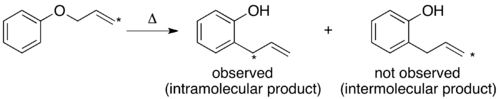
The mechanism of the thermal Claisen rearrangement has been studied by crossover experiment and serves as an excellent example of how to apply this technique.[9] Before the mechanism was determined, it was proposed that the reaction could proceed via an intermolecular or intramolecular route.[1]
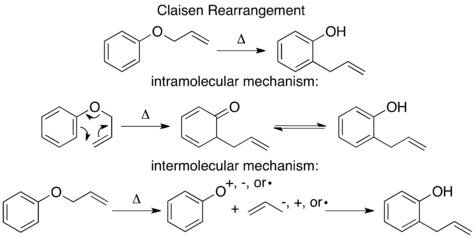
Looking at these two proposed mechanisms, it is clear that a crossover experiment will be suitable for distinguishing between them, as is generally the case for inter- and intramolecular mechanisms. The next step in crossover experiment design is to propose labeled reactants. For a non-isotopic labeling method the smallest perturbation to the system will be by addition of a methyl group at an unreactive position.
Predicting the products given by each mechanism will show whether or not a given crossover experiment design can distinguish between the mechanisms in question. This is particularly relevant when employing an isotopic label. It is possible that labeling at one position could distinguish between only two of several possible mechanisms, while placing the isotopic label at a different position could distinguish between three potential mechanisms or provide insight into transition states or intermediates, etc. After the interpretational value is established it is relevant to consider the practical aspects, such as whether or not the synthesis of the proposed reactant is possible, and how easy or difficult it is to distinguish the predicted products for each proposed mechanism and starting materials.
For the Claisen rearrangement, labeling by addition of a single methyl group produces an under-labeled system. The resulting crossover experiment would not be useful as a mechanistic study since the products of an intermolecular or intramolecular mechanism are identical.
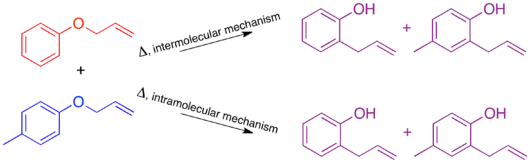
To have a sufficiently labeled system, both “halves” of the molecule that would separate in an intermolecular mechanism must be labeled. This is known as a doubly labeled system, and is generally the requirement for a crossover experiment.[1] Predicting the products of each mechanism then shows that the crossover products are distinct from the non-crossover products. Once this has been established and the products predicted, the experiment can be carried out and the products characterized. When isotopic labeling is used the products are often more varied and the distribution of the label more convoluted. In this case it is also important to explicitly predict the relative amounts of the label expected to appear at each position depending on the mechanism.
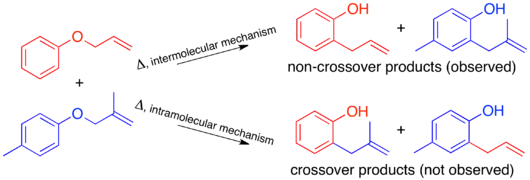
When the crossover experiment on the Claisen rearrangement is carried out only non-crossover products are observed. Based on this the mechanism is determined to be intramolecular, as depicted in the standard arrow-pushing mechanism for this rearrangement.[1][10]
Isotopic labeling experiment
An isotopic labeling experiment is an experiment used in mechanistic study that employs isotopes as labels and traces these labels in the products. Isotopic labeling experiments are commonly considered to be a type of crossover experiment.[1] However, there are far more possibilities for the manner of labeling and potential products in an isotopic labeling experiment than in a traditional crossover experiment. The classification of an isotopic labeling experiment as a crossover experiment is based on the similar underlying concept, goal, and design principles in the two experiments rather than on direct similarity. An isotopic labeling experiment can be designed to be directly analogous to a traditional crossover experiment, but there are many additional ways of carrying out isotopic labeling experiments.
Although isotopic labeling experiments have the advantage of using the smallest perturbation to the reaction system, they are limited by the possibility of isotopic exchange with solvent or other species present in the reaction mixture. If the isotopic label exchanges with another isotope of the same atom in the solvent, the results of an isotopic labeling experiment are unusable. This limits the use of deuterium labeling at certain positions in protic solvents, for example. However, this exchange can be useful when investigating interaction with the solvent of a reaction, since isotopic labeling can detect this interaction.
Isotopic labeling experiments have been carried out on the thermal Claisen rearrangement. When the terminal carbon is labeled with 14C, there is only one product, with the isotopic label appearing at the benzylic position. Since the expected product of an intermolecular mechanism is not observed the conclusion matches that of the traditional crossover experiment.[1][10]
Characterization
A major advantage of the crossover experiment is that the results of the experiment are obtained by direct characterization of the product. The techniques involved are therefore those already familiar to the experimental chemist. Mass spectrometry and NMR spectroscopy are the two most common ways of determining the products and their relative ratios. NMR spectroscopy is particularly useful for isotopic labeling studies that use isotopes of hydrogen or carbon.
IR spectroscopy can be useful in specialized situations, such as when 13CO was used to probe the mechanism of alkyl insertion into metal-carbon monoxide bonds to form metal-acyl complexes. Tracking the 13CO in the products was accomplished using IR spectroscopy because the greater mass of 13C compared to 12C produces a distinctive shift of the ν(CO) stretching frequency to lower energy.[4]
Interpretation
The products that are expected from any given mechanism are determined during the design of the crossover experiment. This can be quite complicated to establish, but it makes for straightforward interpretation of the results. If the observed products match those predicted by a given mechanism, then it is reasonable to conclude that mechanism is operating in the reaction. If the results do not match any expected distribution, it is necessary to consider alternate mechanisms and/or the possibility that the labeling has affected the way the reaction proceeds.
For crossover experiments used to distinguish between intermolecular and intramolecular reactions, the absence of crossover products is less conclusive than the presence of crossover products. This is because solvent cage effects could be masking an intermolecular mechanism.[1][5]
Limitations
Crossover experiments have several limitations. Although useful for distinguishing between proposed reaction mechanisms, they are limited in their ability to provide insight into a mechanism beyond what has already been proposed. The design of a useful crossover experiment relies on having a proposed mechanism on which to base predictions of the label distribution in the products. If the results do not match any expected outcome, the actual mechanism is not obvious from the crossover experiment results. An additional limitation is of course that some systems are just not suitable for crossover experiments. This could be the case if addition of a label alters the mechanism or stops the reaction entirely, if there is no proposed mechanism, if isotopic labels exchange with solvent molecules, or if it is not feasible to synthesize the labeled species necessary for a crossover experiment.
Solvent cage effect
One of the major limitations of the crossover experiment is that it cannot rule out the possibility that solvent cage effects are masking a dissociation mechanism. If crossover products are observed, the evidence that the mechanism cannot be purely intramolecular is conclusive. However, a lack of crossover products is not conclusive evidence that the mechanism is solely intramolecular. Provided that the reaction is carried out in solvent, it is always possible that solvent cage effects are preventing the formation of crossover products.[1][5]
When a molecule is dissolved in a solvent it is appropriate to view the solvent as creating a “cage” around the molecule. The amount of time it takes a given molecule to “escape” this solvent cage varies with the size of the molecule and the strength of the intermolecular forces of the solvent, but is considered to be on the order of 1 x 10−10 seconds.[11] If a reaction occurs faster than the molecules are able to escape the solvent cage, then only non-crossover products will be observed, masking the true reaction mechanism.[5]
When the timescale of the reaction is much slower than the timescale of the solvent cage effect, dissociated species are able to escape the solvent cage and form crossover products. This is an appropriate representation of a reaction in a crossover experiment occurring via an intermolecular mechanism and forming crossover products as expected.
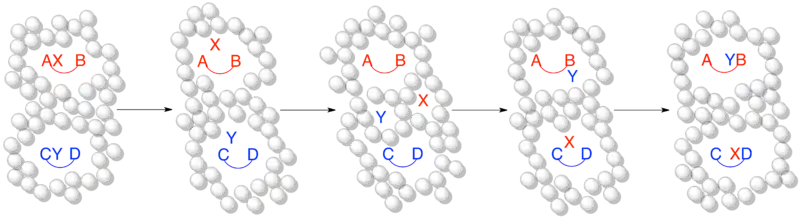
When the timescale of the reaction is faster than or on the same order as the timescale of the solvent cage effect this is a more accurate representation of the same crossover experiment as above. Although a dissociative or intermolecular mechanism is occurring, no crossover occurs because the timescale of the reaction is sufficiently short that the dissociated fragment remains trapped within the solvent cage.
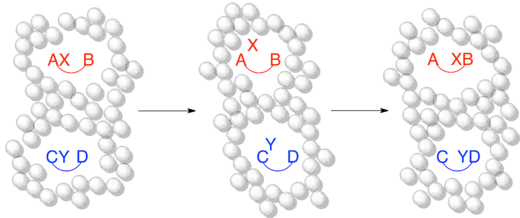
The effect of the solvent cage on crossover experiments is not a purely theoretical concept. One of the first pieces of experimental evidence for the existence of the solvent cage was the observation of the solvent cage effect on a crossover experiment. Since radical recombinations occur on very short timescales compared to non-radical reactions, the solvent cage effect is particularly relevant to radical chemistry.[5] Lyons and Levy were the first to demonstrate the effect of the solvent cage on a radical crossover experiment. When protio- and deutero-azomethane are combined and irradiated in the gas phase, the result is a statistical mixture of the expected non-crossover and crossover radical recombination products, C2H6, CH3CD3, and C2D6, as 1:2:1.[12]

However, when the same reaction is carried out in an isooctane solution, the amount of CH3CD3 formed amounts to less than 0.3% of the total amount of C2H6 formed.[12][13] This demonstrated that the solvent cage effect is capable of significantly altering the results of a crossover experiment, especially on short-timescale reactions such as those involving radicals.
Endocyclic restriction test
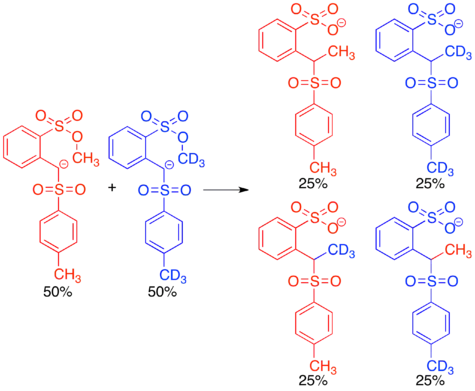
The first endocyclic restriction test was a crossover experiment published by Albert Eschenmoser in 1970. Methylation reactions in which a sulfonyl anion acts as a nucleophile and a methyl (arenesulfonate) serves as an electrophile were known to occur, but it was proposed that they could proceed either intermolecularly or intramolecularly.[2]
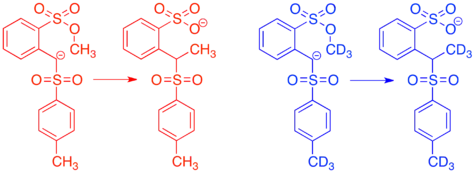
Reacting the protio and doubly labeled deutero sulfonyl anions simultaneously in a crossover experiment gave a 1:1:1:1 mixture of crossover and non-crossover products, clearly indicating that the reaction proceeds via an intermolecular mechanism. This result was surprising, since the intramolecular mechanism would proceed through a cyclic transition state resembling a six-membered ring, which is known to be a favored transition state in many organic mechanisms. The fact that this reaction proceeds via in inter- rather than intramolecular mechanism lead to the conclusion that there are certain restrictions on the geometry of nucleophilic attack in SN2 reactions.[2][14] This concept has been further explored in many subsequent endocyclic restriction tests.[15]
Inorganic chemistry
Mechanisms in inorganic and organometallic chemistry are often complicated and difficult to determine experimentally. Catalytic mechanisms are particularly challenging to study in cases where no metal complex at all aside from the pre-catalyst can be isolated. In the 2013 themed issue of Dalton Transactions entitled “Mechanistic Organometallic Chemistry,” guest editor Robert H. Crabtree recounts a story in which at the midpoint of 20th century the founder of metal carbonyl hydride chemistry referred to organometallic mechanisms as “chemical philosophy.”[8] The themed issue goes on to present seventeen examples of modern mechanistic studies of organometallic reactions. In many cases, crossover experiments, isotope scrambling experiments, kinetic isotope effects, and computational studies are used in conjunction to clarify even a few aspects of an organometallic mechanism.
Crossover experiments provide such uniquely useful insight into inorganic mechanisms that on occasion unusual isotopes are employed for an essential crossover experiment. In the work of E.L. Muetterties on dirhenium decacarbonyl, a crossover experiment was carried out using 185Re and 187Re to determine the mechanism of substitution reactions of rhenium carbonyl dimers. Mass spectrometry was used to distinguish between these isotopes in the products. In the same study, crossover experiments were also carried out using 13CO and 12CO.[16] Isotopic enrichment from an initial isotopic distribution of 63Cu and 65Cu was studied in isotope crossover experiments recently carried out by V.V. Fokin on copper(I)-catalyzed azide-alkyne cycloadditions. The results of these experiments lead to the conclusion that the catalytic cycle of this important click reaction involves a dinuclear copper intermediate.[17]
Reductive elimination
Reductive elimination is a common step in organometallic reaction mechanisms, and particularly in catalytic cycles. In catalytic cycles that form C-H or C-C bonds reductive elimination is often the final product-forming step.[18] Square planar d8 metal complexes are often the active catalysts in C-H or C-C bond forming reactions, and reductive elimination from these species is well understood. There are several known mechanisms for reductive elimination from square planar d8 complexes. In a dissociative mechanism one ligand dissociates and reductive elimination occurs from a three-coordinate intermediate. In a nondissociative mechanism reductive elimination occurs from the square planar complex itself. The ligands undergoing reductive elimination must be cis to each other or otherwise must rearrange to be cis before they can reductively eliminate. Finally, in an associative mechanism a fifth ligand associates and reductive elimination occurs between two adjacent groups on the resulting square pyramidal complex.[4]
Regardless of the specific mechanism, it is consistently true that reductive elimination is an intramolecular process that couples two adjacent ligands. Although this may now seem obvious, when organometallic mechanisms were first being studied there was no proof of these restrictions. A series of crossover experiments experiment reported by J. Stille were among the first experiments to demonstrate that reductive elimination is an intramolecular process and that non-adjacent groups do not reductively eliminate.[4][19][20] Several square planar d8 palladium species were used in the study, with each one having two bound phosphine ligands and two bound methyl groups. One complex, Pd(dppe)(CH3)2, was locked in a cis-confirmation by the chelating phosphine 1,2-bis(diphenylphosphino)ethane (dppe). A second complex, Pd(transphos)(CH3)2 was locked in a trans-confirmation by “transphos,” a chelating phosphine with a rigid aromatic linker.[19]
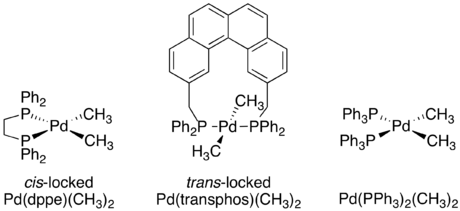
The complexes with cis-methyl groups were already known to undergo reductive elimination to form ethane. A crossover experiment was performed on both Pd(dppe)(CH3)2 and Pd(PPh3)2(CH3)2. In both cases no crossover products were observed, proving the intramolecular nature of reductive elimination.[19]
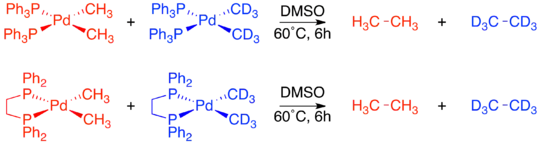
Unlike the two cis-confirmation complexes, Pd(transphos)(CH3)2 did not undergo reductive elimination even when heated to 100 °C. However, addition of methyl iodide to Pd(transphos)(CH3)2 immediately produced ethane. To determine whether or not this reductive elimination was also constrained to only adjacent ligands, an isotope labeling experiment was carried out. The only product was the deuterium-labeled product of cis-elimination. This led to the final conclusion that only ligands adjacent to each other on the metal complex are capable of reductively eliminating.[4][19]

This study also tracked and analyzed reaction rate data, demonstrating the value of employing multiple strategies in a concerted effort to gain as much information as possible about a chemical process. Among other rate experiments, the reaction rates of the cis-trans isomerism were followed as solvent and concentration of excess phosphine ligand were varied. These results were used to establish a mechanism for this isomerization in square planar d8 palladium species that consists of solvent or phosphine association followed by pseudorotation and subsequent dissociation of the solvent or phosphine.[19]
Biochemistry
The mechanisms of enzyme-catalyzed reactions can also be studied using crossover experiments. Examples of the application of this technique in biochemistry include the study of reactions catalyzed by nucleoside diphosphohexose-4,6-dehydratases, the aconitase-catalyzed elimination of water from citrate, and various reactions catalyzed by coenzyme B12-dependent enzymes, among others. Unlike isotope labeling studies in organic and organometallic chemistry, which typically use deuterium when an isotope of hydrogen is desired, biochemical crossover experiments frequently employ tritium.[21] This is due to the fact that tritium is radioactive and can be tracked using the autoradiographs of gels in gel electrophoresis.
Mechanism of aconitase action
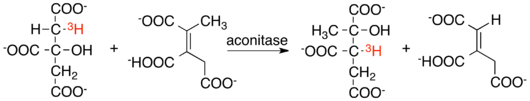
Isotope labeling experiments and crossover experiments were essential to early efforts to understand the mechanism of aconitase action. Isotope scrambling experiments using tritium, deuterium, and 18O were carried out on the aconitase hydratase reaction by I.A. Rose and E.L. O'Connell.[22] Using the results of these experiments it was possible to construct a general mechanism for the reaction. Further work has been done to refine this mechanism since these early experiments.[23][24]
One such isotope scrambling experiment was the reaction of [2R-3H]citrate with aconitase in the presence of 2-methyl-cis-aconitate. This reaction produced both unlabeled cis-aconitate and 2-methyl-[3-3H]isocitrate. The ability of the reaction to produce intermolecular transfer of tritium at this position indicates that the proton removed from citrate does not exchange with solvent. A similar experiment reacting [2-18OH]isocitrate with aconitase failed to produce isotopically labeled citrate, demonstrating that the hydroxyl group, unlike the removed proton, exchanges with solvent every turnover.[21][22]
References
- Carroll, Felix A.; Perspectives on Structure and Mechanism in Organic Chemistry; Brooks/Cole Publishing, Pacific Grove, CA, 1998.
- Brucker, Reinhard; Advanced Organic Chemistry: Reaction Mechanisms; Academic Press, San Diego, 2002.
- http://www.chemgapedia.de/vsengine/vlu/vsc/en/ch/12/oc/vlu_organik/aufklaerung/aufklaerung_e_kreuz_kinetik.vlu.html
- Crabtree, Robert H.; The Organometallic Chemistry of the Transition Metals; Third Ed. Wiley, NY, 2001.
- Edwards, John O., Ed; Progress in Inorganic Chemistry: Inorganic Reaction Mechanisms; vol. 17, 1972.
- Xiaoping Sun (5 June 2013). Organic Mechanisms: Reactions, Methodology, and Biological Applications. John Wiley & Sons. ISBN 978-1-118-50791-9.
- Eldik,R. Chem. Rev. 2005, 105, 1917.
- Crabtree, R. H.; Dalton Trans., 2013, 42, 4104.
- Aditi Sangal. Krishna's Advanced Organic Chemistry; Volume 1. Krishna Prakashan Media. ISBN 978-81-8283-078-3.
- Francis A. Carey; Richard J. Sundberg (13 June 2007). Advanced Organic Chemistry: Part A: Structure and Mechanisms. Springer. ISBN 978-0-387-44897-8.
- Horspool, William M.; Lenci, Francesco, Eds; CRC Handbook of Organic Photochemistry and Photobiology; CRC Press, 2004.
- Lyon, R.K.; Levy, D.H. J. Am. Chem. Soc. 1961, 83, 4290.
- Denisov, E.T.; Denisova, T. G.; Pokidova, T.S.; Handbook of Free Radical Initiators; John Wiley & Sons, Hoboken, N.J., 2003.
- Tenud, L.; Farouq, S.; Seible, J.; Eschenmoser, A. Helv. Chim. Acta 1970, 53, 2059.
- Beak, P. Acc. Chem. Res. 1992, 25, 215.
- Stolzenberg, A. M.; E. L. Muetterties. J. Am. Chem. Soc. 1983, 105.4, 822.
- Worrell, B.T.; Malik, J.A.; Fokin, V.V. Science 2013, 340, 457.
- Royal Society of Chemistry (Great Britain). Faraday Division (2003). Quantum Inorganic Chemistry. Royal Society of Chemistry. ISBN 978-0-85404-967-7.
- Gillie, S. J. Am. Chem. Soc. 1980, 102, 4933.
- http://www.ilpi.com/organomet/reductive.html
- Silverman, Richard B.; The Organic Chemistry of Enzyme-Catalyzed Reactions; Academic Press, London, 2002.
- Rose, I.A.; O'Connell, E.L. J. Biol. Chem. 1967, 242, 870. http://www.jbc.org/content/242/8/1870.long
- Glusker, J.P. J. Mol. Biol. 1968, 38, 149.
- Villafranca, J.J. J. Biol. Chem. 1971, 249, 6149.