Intramolecular reaction
Intramolecular in chemistry describes a process or characteristic limited within the structure of a single molecule, a property or phenomenon limited to the extent of a single molecule.
Examples
- intramolecular hydride transfer (transfer of a hydride ion from one part to another within the same molecule)
- intramolecular hydrogen bond (a hydrogen bond formed between two functional groups of the same molecule)
- cyclization of ω-haloalkylamines and alcohols to form the corresponding saturated nitrogen and oxygen heterocycles, respectively (an SN2 reaction within the same molecule)
In intramolecular organic reactions, two reaction sites are contained within a single molecule. This creates a very high effective concentration (resulting in high reaction rates), and, therefore, many intramolecular reactions that would not occur as an intermolecular reaction between two compounds take place.
Examples of intramolecular reactions are the Smiles rearrangement, the Dieckmann condensation and the Madelung synthesis.
Relative rates
Intramolecular reactions, especially ones leading to the formation of 5- and 6-membered rings, are rapid compared to an analogous intermolecular process. This is largely a consequence of the reduced entropic cost for reaching the transition state of ring formation and the absence of significant strain associated with formation of rings of these sizes. For the formation of different ring sizes via cyclization of substrates of varying tether length, the order of reaction rates (rate constants kn for the formation of an n-membered ring) is usually k5 > k6 > k3 > k7 > k4 as shown below for a series of ω-bromoalkylamines. This somewhat complicated rate trend reflects the interplay of these entropic and strain factors:
| n | krel | n | krel | n | krel |
|---|---|---|---|---|---|
| 3 | 0.1 | 6 | 1.7 | 12 | 0.00001 |
| 4 | 0.002 | 7 | 0.03 | 14 | 0.0003 |
| 5 | 100 | 10 | 0.00000001 | 15 | 0.0003 |
For the 'small rings' (3- and 4- membered), the slow rates is a consequence of angle strain experienced at the transition state. Although three-membered rings are more strained, formation of aziridine is faster than formation of azetidine due to the proximity of the leaving group and nucleophile in the former, which increases the probability that they would meet in a reactive conformation. The same reasoning holds for the 'unstrained rings' (5-, 6-, and 7-membered). The formation of 'medium-sized rings' (8- to 13-membered) is particularly disfavorable due to a combination of an increasingly unfavorable entropic cost and the additional presence of transannular strain arising from steric interactions across the ring. Finally, for 'large rings' (14-membered or higher), the rate constants level off, as the distance between the leaving group and nucleophile is now so large the reaction is now effectively intermolecular.[1][2]
Although the details may change somewhat, the general trends hold for a variety of intramolecular reactions, including radical-mediated and (in some cases) transition metal-catalyzed processes.
Tethered intramolecular [2+2] reactions
Tethered intramolecular [2+2] reactions entail the formation of cyclobutane and cyclobutanone via intramolecular 2+2 photocycloadditions. Tethering ensures formation of a multi-cyclic system.

The length of the tether affects the stereochemical outcome of the [2+2] reaction. Longer tethers tend to generate the "straight" product where the terminal carbon of the alkene is linked to the -carbon of the enone.[3] When the tether consists only two carbons, the “bent” product is generated where the -carbon of the enone is connected to the terminal carbon of the alkene (Figure 2).[4]



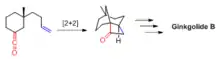
Tethered [2+2] reactions have been used to synthesize organic compounds with interesting ring systems and topologies. For example, [2+2] photocyclization was used to construct the tricyclic core structure in ginkgolide B by E. J. Corey and co-workers in 1988.[5]
Molecular tethers
In a niche concept called molecular tethers, otherwise-intermolecular reactions can be made temporarily intramolecular by anchoring both reactions by a tether with all the advantages associated to it. Popular choices of tether contain a carbonate ester, boronic ester, silyl ether, or a silyl acetal link (silicon tethers)[6][7] which are fairly inert in many organic reactions yet can be cleaved by specific reagents. The main hurdle for this strategy to work is selecting the proper length for the tether and making sure reactive groups have an optimal orientation with respect to each other. An examples is a Pauson–Khand reaction of an alkene and an alkyne tethered together via a silyl ether.[8]

In this particular reaction, the tether angle bringing the reactive groups together is effectively reduced by placing isopropyl groups on the silicon atom via the Thorpe–Ingold effect. No reaction takes place when these bulky groups are replaced by smaller methyl groups.
Another example is a photochemical [2+2]cycloaddition with two alkene groups tethered through a silicon acetal group (racemic, the other enantiomer not depicted), which is subsequently cleaved by TBAF yielding the endo-diol.

Without the tether, the exo isomer forms.[9]
References
- Streitwieser, Andrew; Heathcock, Clayton H.; Kosower, Edward M. (2017). Introduction to Organic Chemistry. New Delhi: Medtech (Scientific International, reprint of 1998 revised 4th edition, Macmillan). p. 198. ISBN 9789385998898.
- Jonathan Clayden (2001). Organic chemistry. Oxford: Oxford University Press. pp. 454]. ISBN 0198503474. OCLC 43338068.
- Coates, R. M.; Senter, P. D.; Baker, W. R. (1982). "Annelative Ring Expansion via Intramolecular [2 + 2] Photocycloaddition of α,β-Unsaturated γ-Lactones and Reductive Cleavage: Synthesis of Hydrocyclopentacyclooctene-5-carboxylates". J. Org. Chem. 47: 3597. doi:10.1021/jo00140a001.CS1 maint: uses authors parameter (link)
- Tamura, Y.; Kita, Y.; Ishibashi, H.; Ikeda, M. (1971). "Intramolecular photocycloaddition of 3-allyloxy- and 3-allylamino-cyclohex-2-enones: formation of oxa- and aza-bicyclo[2,1,1]hexanes". J. Chem. Soc. D. 19: 1167. doi:10.1039/C29710001167.
- Corey, E. J.; Kang, M. C.; Desai, M. C.; Ghosh, A. K.; Houpis, I. N. (1988). "Total Synthesis of (.+-.)-Ginkgolide B". J. Am. Chem. Soc. 110: 649–651. doi:10.1021/ja00210a083. PMC 6746322. PMID 31527923.CS1 maint: uses authors parameter (link)
- Cox, Liam R.; Ley, Steven V. (2007). "Use of the Temporary Connection in Organic Synthesis". In Diederich, François; Stang, Peter J. (eds.). Templated Organic Synthesis. pp. 274–395. doi:10.1002/9783527613526.
- Bracegirdle, S.; Anderson, E. A. (2010). "Recent advances in the use of temporary silicon tethers in metal-mediated reactions". Chem. Soc. Rev. 39: 4114–4129. doi:10.1039/C0CS00007H.
- Dobbs, A.; Miller, I.; Martinovic, S. (2007). "The use of silicon-based tethers for the Pauson-Khand reaction". Beilstein Journal of Organic Chemistry. 2007 (3): 21. doi:10.1186/1860-5397-3-21. PMC 1949821. PMID 17617903.
- Booker-Milburn, Kevin I.; Gulten, Sirin; Sharpe, Andrew (1997). "Diastereoselective intramolecular photochemical [2 + 2] cycloaddition reactions of tethered l-(+)-valinol derived tetrahydrophthalimides". Chem. Commun. 1997: 1385–1386. doi:10.1039/a702386c.