Dihydroxylation
Dihydroxylation is the process by which an alkene is converted into a vicinal diol. Although there are many routes to accomplish this oxidation, the most common and direct processes use a high-oxidation-state transition metal (typically osmium or manganese). The metal is often used as a catalyst, with some other stoichiometric oxidant present.[1] In addition, other transition metals and non-transition metal methods have been developed and used to catalyze the reaction.

Mechanism
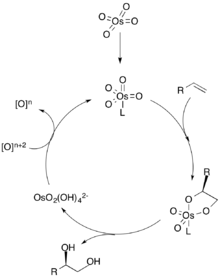
In the dihydroxylation mechanism, a ligand first coordinates to the metal catalyst (depicted as osmium), which dictates the chiral selectivity of the olefin. The alkene then coordinates to the metal through a [3+2] cycloaddition, and the ligand dissociates from the metal catalyst. Hydrolysis of the olefin then yields the vicinal diol, and oxidation of the catalyst by a stoichiometric oxidant regenerates the metal catalyst to repeat the cycle.[2] The concentration of the olefin is crucial to the enantiomeric excess of the diol since higher concentrations of the alkene can associate with the other catalytic site to produce the other enantiomer.[3]
Osmium catalyzed reactions
Osmium tetroxide (OsO4) is a popular oxidant used in the dihydroxylation of alkenes because of its reliability and efficiency with producing syn-diols. Since it is expensive and toxic, catalytic amounts of OsO4 are used in conjunction with a stoichiometric oxidizing agent.[2][3] The Milas hydroxylation, Upjohn dihydroxylation, and Sharpless asymmetric dihydroxylation reactions all use osmium as the catalyst as well as varying secondary oxidizing agents.
Milas
The Milas dihydroxylation was introduced in 1930, and uses hydrogen peroxide as the stoichiometric oxidizing agent.[4] Although the method can produce diols, overoxidation to the dicarbonyl compound has led to difficulties isolating the vicinal diol.[4] Therefore, the Milas protocol has been replaced by the Upjohn and Sharpless asymmetric dihydroxylation.
Upjohn
Upjohn dihydroxylation was reported in 1973 and uses OsO4 as the active catalyst in the dihydroxylation procedure. It also employs N-Methylmorpholine N-oxide (NMO) as the stoichiometric oxidant to regenerate the osmium catalyst, allowing for catalytic amounts of osmium to be used.[2][5] The Upjohn protocol yields high conversions to the vicinal diol and tolerates many substrates. However, the protocol cannot dihydroxylate tetrasubstituted alkenes.[2] The Upjohn conditions can be used for synthesizing anti-diols from allylic alcohols, as demonstrated by Kishi and coworkers.[6]
Sharpless asymmetric
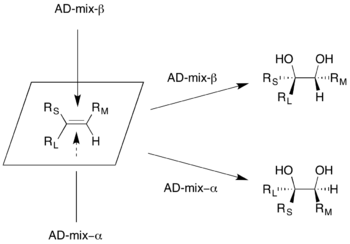
The Sharpless asymmetric dihydroxylation was developed by K. Barry Sharpless to use catalytic amounts of OsO4 along with the stoichiometric oxidant K3[Fe(CN)6].[1][2][7] The reaction is performed in the presence of a chiral auxiliary. The selection of dihydroquinidine (DHQD) or dihydroquinine (DHQ) as a chiral auxiliary dictates the facial selectivity of the olefin, since the absolute configuration of the ligands are opposite.[2][7][8] The catalyst, oxidant, and chiral auxiliary can be purchased premixed for selective dihydroxylation. AD-mix-α contains the chiral auxiliary (DHQ)2PHAL, which positions OsO4 on the alpha-face of the olefin; AD-mix-β contains (DHQD)2PHAL and delivers hydroxyl groups to the beta-face.[1][9] The Sharpless asymmetric dihydroxylation has a large scope for substrate selectivity by changing the chiral auxiliary class.[7]
Other variants
As mentioned above, the ability to synthesize anti-diols from allylic alcohols can be achieved with the use of NMO as a stoichiometric oxidant.[6] The use of tetramethylenediamine (TMEDA) as a ligand produced syn-diols with a favorable diastereomeric ratio compared to Kishi’s protocol; however, stoichiometric osmium is employed. Syn-selectivity is due to the hydrogen bond donor ability of the allylic alcohol and the acceptor ability of the diamine.[10][11][12] This has since been applied to homoallylic systems. [13]
Other dihydroxylation methods
Since osmium tetroxide is expensive and toxic, other metals have been used to prepare vicinal diols from olefins. Another popular metal used in dihydroxylation is ruthenium. Although it is highly oxidative, ruthenium has been used because of its short reaction time and its cost-effectiveness.[14] Typically, the ruthenium tetroxide is created in situ from ruthenium trichloride, and a secondary oxidant NaIO4 is used to regenerate the catalyst. The turnover-limiting step of the reaction is the hydrolysis step; therefore, sulfuric acid is added to increase the rate of this step.[14][15]
Manganese is also used in dihydroxylation and is often chosen when osmium tetroxide methods yield poor results.[15] Similar to ruthenium, the oxidation potential of manganese is high, leading to over-oxidation of substrates. Potassium permanganate is often used as the oxidant for dihydroxylation; however, due to its poor solubility in organic solvent, a phase-transfer catalyst (such as benzyltriethylammonium chloride, TEBACl) is also added to increase the number of substrates for dihydroxylation.[15]
Prévost and Woodward dihydroxylation
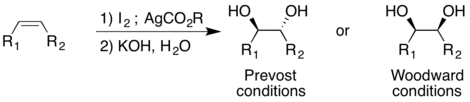
Unlike the other methods described that use transition metals as catalyst, the Prévost and Woodward methods use iodine and a silver salt. However, the addition of water into the reaction directs the cis- and trans- addition of the hydroxyl groups. The Prévost reaction typically uses silver benzoate to produce trans-diols; the Woodward modification of the Prévost reaction uses silver acetate to produce cis-diols. In both the Prévost and Woodward reactions, iodine is first added to the alkene producing a cyclic iodinium ion. The anion from the corresponding silver salt is then added by nucleophilic substitution to the iodinium ion.[16]
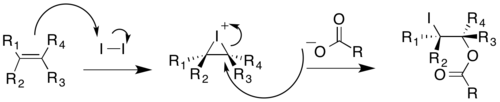
In the Prévost reaction, the iodinium ion undergoes nucleophilic attack by benzoate anion. The benzoate anion acts as a nucleophile again to displace iodide through a neighboring-group participation mechanism. A second benzoate anion reacts with the intermediate to produce the anti-substituted dibenzoate product, which can then undergo hydrolysis to yield trans-diols.[16]
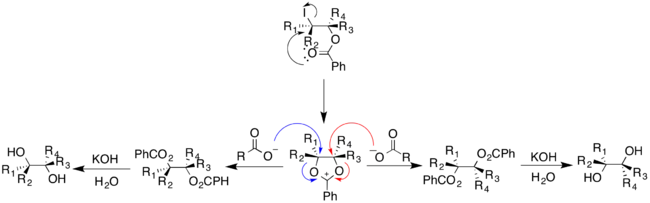
The Woodward modification of the Prévost reaction yields cis-diols. Acetate anion reacts with the cyclic iodinium ion to yield an oxonium ion intermediate. This can then readily react with water to give the monoacetate, which can then be hydrolyzed to give a cis-diol [17]
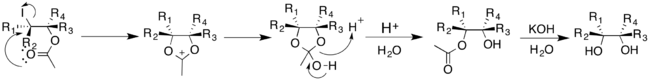
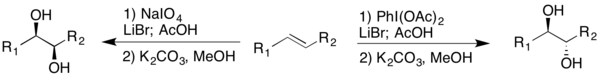
To eliminate the need for silver salts, Sudalai and coworkers modified the Prévost-Woodward reaction; the reaction is catalyzed with LiBr, and uses NaIO4 and PhI(OAc)2 as oxidants.[18] LiBr reacts with NaIO4 and acetic acid to produce lithium acetate, which can then proceed through the reaction as previously mentioned. The protocol produced high dr for the corresponding diol, depending on the oxidant chosen.
Applications
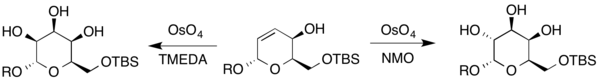
The synthesis of highly substituted and stereospecific sugars is important since polysaccharides make up a large class of compounds found in nature. One specific example is in the biologically active molecule kakelokelose, which has been shown to have anti-HIV activity.[19] Research conducted by Harris et al. have worked on an enantiospecific synthesis of sugars pertaining to kakelokelose and other sugars, employing many different dihydroxylation reactions with osmium catalyst. Vinylfuran was reacted under Sharpless conditions with AD-mix-α to yield (R)-diol. Later, a resulting dihydropyran was reacted under Upjohn conditions to yield the resulting sugar, mannose (where R represents either H or a protecting group).[19]

Additionally, talose and gulose were also synthesized from a different dihydropyran. Since the compound contains an allylic alcohol, Upjohn conditions and the Upjohn modification using TMEDA as the secondary oxidant to create the resulting sugars (where R represents either H or a protecting group).[19]
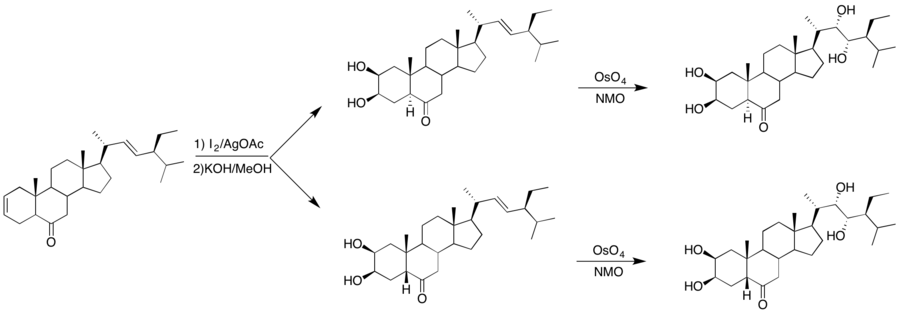
Another application of dihydroxylation methods is in the synthesis of steroids. Brassinosteroids are a class of steroids shown to regulate plant growth and has been shown to have agricultural activity as an insecticide. This class of steroids contains the standard framework of steroids in addition to four vicinal diols that have their own stereochemistry.[20] Brosa installed the hydroxyl groups in the steroid using both Woodward conditions to yield a cis-diol to the A ring of the steroid. Then, the alkene chain on the D ring was dihydroxylated to yield the second cis-diol using OsO4 and NMO as the stoichiometric oxidant.[21]
References
- Carey, Francis A.; Sundberg, Richard J. Advanced Organic Chemistry Part B: Reactions and Synthesis (5th ed.). Springer.
- Schroder, M. (1980). "Osmium tetraoxide cis hydroxylation of unsaturated substrates". Chem. Rev. 80 (2): 187–213. doi:10.1021/cr60324a003.
- Kolbe, H.C.; VanNieuwanhze, M.S.; Sharpless, K.B. (1994). "Catalytic Asymmetric Dihydroxylation". Chem. Rev. 94 (8): 2483–2547. doi:10.1021/cr00032a009.
- Milas, N.A.; Sussman, S. (1936). "The Hydroxylation of the Double Bond 1". J. Am. Chem. Soc. 58 (7): 1302–4. doi:10.1021/ja01298a065.
- Dupau, P.; Epple, R.; Thomas, A.A.; Fokin, V.V.; Sharpless, K.B. (2002). "Osmium-Catalyzed Dihydroxylation of Olefins in Acidic Media: Old Process, New Tricks". Adv. Synth. Catal. 344 (3–4): 421–33. doi:10.1002/1615-4169(200206)344:3/4<421::AID-ADSC421>3.0.CO;2-F.
- Cha, J.K.; Christ, W.J.; Kishi, Y. (1983). "1983". Tetrahedron Lett. 24: 3943–6. doi:10.1016/s0040-4039(00)88231-3.
- Morikawa, K.; Park, J.; Anderson, P.G.; Hashiyama, T.; Sharpless, K.B. (1993). "Catalytic asymmetric dihydroxylation of tetrasubstituted olefins". J. Am. Chem. Soc. 115 (18): 8463–4. doi:10.1021/ja00071a072.
- Carey, F.A.; Sundberg, R.J. (2007). Advanced Organic Chemistry Part A: Structure and Mechanisms. Springer. p. 202.
- Xu, D.X.; Crispino, G.A.; Sharpless, K.B. (1992). "Selective asymmetric dihydroxylation (AD) of dienes". J. Am. Chem. Soc. 114 (19): 7570–1. doi:10.1021/ja00045a043.
- Donohoe, T.J.; Blades, K.; Moore, P.R.; Waring, M.J.; Winter, J.J.G.; Helliwell, M.; Newcombe, N.J.; Stemp, G. (2002). "Directed dihydroxylation of cyclic allylic alcohols and trichloroacetamides using OsO4/TMEDA". J. Org. Chem. 67 (23): 7946–56. doi:10.1021/jo026161y. PMID 12423122.
- Donohoe, T.J.; Moore, P.R.; Waring, M.J.; Newcombe, Nicholas J. (1997). "The directed dihydroxylation of allylic alcohols". Tetrahedron Lett. 38 (28): 5027–30. doi:10.1016/s0040-4039(97)01061-7.
- Donohoe, T.J. (2002). "Development of the Directed Dihydroxylation Reaction". Synlett (8): 1223–32. doi:10.1055/s-2002-32947.
- Donohoe, Timothy J.; Mitchell, Lee; Waring, Michael J.; Helliwell, Madeleine; Bell, Andrew; Newcombe, Nicholas J. (2003-06-10). "Scope of the directed dihydroxylation: application to cyclic homoallylic alcohols and trihaloacetamides". Organic & Biomolecular Chemistry. 1 (12): 2173–2186. doi:10.1039/B303081D. ISSN 1477-0539. PMID 12945911.
- Plietker, B.; Niggemann, M. (2003). "An improved protocol for the RuO4-catalyzed dihydroxylation of olefins". Org. Lett. 5 (18): 3353–6. doi:10.1021/ol035335a. PMID 12943425.
- Bataille, C.J.R.; Donohoe, T.J. (2011). "Osmium-free direct syn-dihydroxylation of alkenes". Chem. Soc. Rev. 40 (1): 114–28. doi:10.1039/b923880h. PMID 21049111.
- Kurti, L.; Czako, B. (2005). Strategic Applications of Named Reactions in Organic Synthesis. Elsevier. pp. 360–1.
- Woodward, R.B.; Brutcher, Jr., F.V. (1958). "cis-Hydroxylation of a Synthetic Steroid Intermediate with Iodine, Silver Acetate and Wet Acetic Acid". J. Am. Chem. Soc. 80: 209–11. doi:10.1021/ja01534a053.
- Emmanuvel, L.; Shaikh, T.M.A.; Sudalai, A. (2005). "NaIO4/Li Br-mediated diastereoselective dihydroxylation of olefins: A catalytic approach to the Prevost-Woodward reaction". Org. Lett. 7 (22): 5071–4. doi:10.1021/ol052080n. PMID 16235960.
- Harris, J.M.; Keranen, M.D.; O'Doherty, G.A. (1999). "Syntheses of D- and L-Mannose, Gulose, and Talose via Diastereoselective and Enantioselective Dihydroxylation Reactions". J. Org. Chem. 64 (9): 2982–3. doi:10.1021/jo990410. PMID 11674384.
- Bishop, G.; Koncz, Csaba (2002). "Brassinosteroids and Plant Steroid Hormone Signaling". The Plant Cell. 14: S97–110. doi:10.1105/tpc.001461. PMC 151250. PMID 12045272.
- Brosa, C.; Nusimovich, S; Peracaula, R (1994). "Synthesis of new brassinosteroids with potential activity as antiecdysteroids". Steroids. 59 (8): 463–7. doi:10.1016/0039-128x(94)90058-2. PMID 7985206. S2CID 45409677.