f-block metallocene
f-Block Metallocene refers to a class of organometallic sandwich compounds with f-Block metal and electron-rich ligands like cyclopentadienyl anion.
History
The first prepared and well-characterized f-block metallocenes were the tris(cyclopentadienyl) lanthanide complexes, (C5H5)3Ln (Ln = La, Ce, Pr, Nd, Sm and Gd).[1][2] However, their significance is limited more to their existences and structures than to their reactivity. The cyclopentadienyl ligands of f-block metallocenes were considered as inert ancillary ligands, only capable of enhancing their stability and solubility, but not their reactivity. In addition, only late and small metals in the lanthanide series, i.e., elements from Sm to Lu, are trivalent metallocene complexes, [(C5H5)2LnZ]n[3][4] In 1980, the pentamethylcyclopentadienyl ligand, C5Me−
5, was introduced to prepare the lanthanide complexes with all metals in the series.[3][5][6][7][8] Apart from improving the stability and solubility of the complexes, it was demonstrated to participate in organometallic reactions. Subsequently, William J. Evans and his coworkers successfully isolated (C5Me5)2Sm(THF)2[8] and (C5Me5)2Sm,[9] making a breakthrough in f-block metallocenes, since both of these two organosamarium(II) complexes were unexpectedly found to participate in the coordination, activation and transformation of a variety of unsaturated compounds, including olefins,[8][10][11][12] dinitrogen,[13] internal alkynens,[14][15] phosphaalkynes,[16] carbon monoxide,[17] carbon dioxide,[18] isonitriles,[19] diazine derivatives,[20][21][22] imines[23] and polycyclic aromatic hydrocarbons (PAHs).[24] Moreover, due to its strong reducing potential, it was used to synthesize [(C5Me5)2Sm(µ-H)]2 and other trivalent f-block element complexes.[14] Subsequently, tris(pentamethylcyclopentadienyl) lanthanide complexes, (C5Me5)3Ln, and their relevant complexes were synthesized from Sm2+ complexes. These metallocenes included (C5Me5)3Sm, [(C5H3(SiMe3)2]3Sm, (C5Me5)2Sm(C5H5), [(C5Me5)2Sm]2(µ-C5H5).[25][26][27] Later, one tris(pentamethylcyclopentadienyl) f-element halide complex, (C5Me5)3UCl, was successfully isolated as the intermediate of the formation of (C5Me5)2UCl2.[28] It is worthy mentioning that (C5Me5)3UCl has a very similar structure as (C5Me5)3U and its uranium-chloride bond (2.90 Å) is relatively longer than the uranium-chloride bonds of other analogues.[29] Its existence also indicates that the larger f-block elements are capable of accommodating additional ligands in addition to the three cyclopentadienyl ligands resulting in the isolation of the following complexes: (C5Me5)3UF,[28] (C5H3(TMS)2)3Th[30] and (C5Me5)3ThH.[31]
Synthesis
I. the synthesis of the first f-block metallocenes is described by following equation:[1][2]

II. Preparation of (C5Me5)3Sm:
(i) the first (C5Me5)3Sm was prepared via exploratory Sm2+ chemistry with cyclooctatetraene:[25]
3Sm_synthetic_method.png.webp)
(ii) Similar to method (i), (C5Me5)3Sm can be efficiently synthesized from a Sm2+ precursor and (C5Me5)2Pb:[32]
3Sm_synthetic_method.png.webp)
In this pathway, (C5Me5)2Sm(OEt2) is used since it is more readily available than (C5Me5)3Sm and does not react THF.
(iii) additionally, (C5Me5)3Sm can also be prepared from trivalent precursors, without ring opening THF.
3Sm_synthetic_method.png.webp)
This solvated cation route generally allows the preparation of all (C5Me5)3Ln complexes since [(C5Me5)3LnH]x is known for all lanthanide elements.
An alternative unsolvated cation pathway prohibits THF during the reaction since (C5Me5)3Sm can ring open THF.[33][34]
3Sm_synthetic_method_(2).png.webp)
III. Generally, in order to synthesize (C5Me5)3M, the starting materials and the reaction conditions require optimizing to ensure (C5Me5)3M is the most favored product.[29] In addition, compounds capable of reacting with (C5Me5)3M, such as THF, nitriles or isolnitriles, should be avoided.[29] Therefore, the following routes are possible options:
(i) For M=Ln including La, Ce, Pr, Nd and Gd, unsolvated cation route is preferred since [(C5Me5)2LnH]x complexes are too reactive.
3M_synthetic_method.png.webp)
Notably, the synthesis of (C5Me5)3La and (C5Me5)3Ce requires the usage of silylated glassware since they are easily oxidized.
(ii) For M=actinide like U, solvated cation route can be used.
3M_synthetic_method.png.webp)
IV. Synthesis of (C5Me5)3MZ with Z=X, H, etc.[28][31]
3MZ_synthetic_method.png.webp)
3MZ_synthetic_method.png.webp)
Reactivity
Unlike d-block elements, f-block elements do not follow 18-electron rule due to their f-orbitals.[35] The following complexes, (C5H4SiMe3)3Ln, have extremely negative reduction potentials of -2.7 to -3.9 Volts versus the standard hydrogen electrode (NHE).[36] Furthermore, in comparison with d-orbitals of transition metals, the radial extension of their 4f-orbitals are really small and limited, which greatly reduces the orbital effects.[37] More specifically, its 4fn electron configurations have almost no effect on its chemical reactivity and its electrostatic interactions require optimizing through ligand geometries. Moreover, the reactivity of the f-block element complexes relies heavily on their sterics. In other words, a sterically saturated structure offers the best stability, and so, both ligand size or metal size can be altered to modify the reactivity.[37] These special properties allow the following reactions to occur.
Alkyl-like Reactivity
Like alkyl group, the electron-rich ligand of f-block metallocenes can act as a nucleophile during organometallic reactions. For example, they can polymerize olefins,[38] and participate in ring opening polymerizations,[34][39] etc.
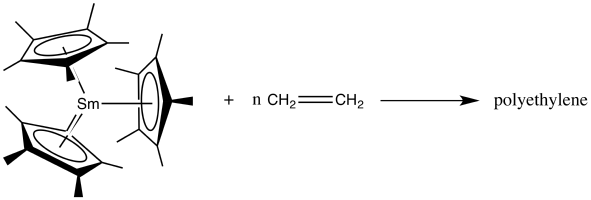
Ligand Cleavage
Especially in the presence of Lewis acids like B(C6F5)3 or Al2Me6, the Cp and other similar ligands can be removed in the following way.[40]
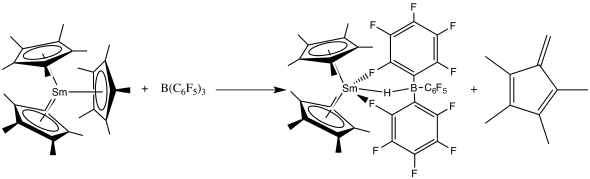
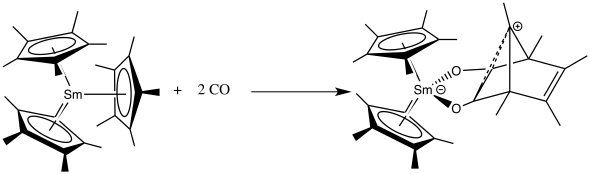
Insertions
The f-block metallocenes are able to undergo insertion reactions of compounds like carbon monoxide,[41] nitriles or isocyanates.[34]
Ordinary reductions
Since f-block metallocenes are very electron-rich, they tend to lose one electron and a pentamethylcyclopentadienyl ligand.

Sterically induced reduction (SIR)
Sterically crowded complexes like (C5Me5)3Sm are able to provide strong reductivity and so this type of reaction was named as SIR.[42] Due to the strong steric hindrance, one ligand cannot bind to the metal center at the ideal distance and so the complex is not stable.[29] Thus, the anion is more inclined to become oxidized and leave the complex, resulting in a highly reducing metal complex.
_of_the_f-block_metallocene.png.webp)
References
- Wilkinson, G.; Birmingham, J. M. J. Am. Chem. Soc. 1954, 76, 6210.
- Birmingham, J. M.; Wilkinson, G. J. Am. Chem. Soc. 1956, 78, 42.
- Evans, W. J.; Wayda, A. L. Inorg. Chem. 1980, 19, 2190.
- Evans, W. J. In The Chemistry of the Metal-Carbon Bond; Hartley, F. R., Patai, S., Eds.; John Wiley: New York, 1982.
- King, R. B. Coord. Chem. Rev. 1976, 20, 155.
- Wolczanski, P. T.; Bercaw, J. E. Acc. Chem. Res. 1980, 11, 121.
- Watson, P. L. J. Chem. Soc., Chem. Commun. 1980, 652.
- Tilley, T. D.; Anderson, R. A.; Spencer, B.; Ruben, H.; Zalkin, A.; Templeton, D. H. Inorg. Chem. 1980, 19, 2999.
- Evans, W. J.; Hughes, L. A.; Hanusa, T. P. J. Am. Chem. Soc. 1984, 106, 4270.
- Evans, W. J.; Ulibarri, T. A.; Ziller, J. W. J. Am. Chem. Soc. 219.
- Evans, W. J.; Ulibarri, T. A.; Ziller, J. W. J. Am. Chem. Soc. 1990, 112, 2314.
- Evans, W. J.; Chamberlain, L. R.; Ulibarri, T. A.; Ziller, J. W. J. Am. Chem. Soc. 1988, 110, 6423.
- Evans, W. J.; Ulibarri, T. A.; Ziller, J. W. J. Am. Chem. Soc. 1988, 110, 6877.
- Evans, W. J.; Bloom, I.; Hunter, W. E.; Atwood, J. L. J. Am. Chem. Soc. 1983, 105, 1401.
- Evans, W. J.; Giarikos, D. G.; Robledo, C. B.; Leong, V. S.; Ziller, J. W. Organometallics 2001, 20, 5648.
- Recknagel, A.; Stalke, D.; Roesky, H. W.; Edelmann, F. T. Angew. Chem. Int. Ed. Engl. 1989, 28, 445.
- Evans, W. J.; Grate, J. W.; Hughes, L. A.; Zhang, H.; Atwood, J. L. J. Am. Chem. Soc. 1985, 107, 3728.
- Evans, W. J.; Seibel, C. A.; Ziller, J. W. Inorg. Chem. 1998, 37, 770.
- Evans, W. J.; Drummond, D. K. Organometallics 1988, 7, 797.
- Evans, W. J.; Drummond, D. K.; Bott, S. G.; Atwood, J. L. Organometallics 1986, 5, 2389.
- Evans, W. J.; Drummond, D. K. J. Am. Chem. Soc. 1986, 108, 7440.
- Evans, W. J.; Drummond, D. K.; Chamberlain, L. R.; Doedens, R. J.; Bott, S. G.; Zhang, H.; Atwood, J. L. J. Am. Chem. Soc. 1988, 110, 4983.
- Evans, W. J.; Drummond, D. K. J. Am. Chem. Soc. 1989, 111, 3329.
- Evans, W. J.; Gonzales, S. L.; Ziller, J. W. J. Am. Chem. Soc. 1994, 116, 2600.
- Evans, W. J.; Gonzales, S. L.; Ziller, J. W. J. Am. Chem. Soc. 1991, 113, 7423.
- Evans, W. J.; Keyer, R. A.; Ziller, J. W. J. Organomet. Chem. 1990, 394, 87.
- Evans, W. J.; Ulibarri, T. A. J. Am. Chem. Soc. 1987, 109, 4292.
- Evans, W. J.; Nyce, G. W.; Johnston, M. A.; Ziller, J. W. J. Am. Chem. Soc. 2000, 122, 12019.
- Evans, W. J.; Davis, B. J. Chem. Rev. 2002, 102, 2119−2136.
- Blake, P. C.; Edelstein, N. M.; Hitchcock, P. B.; Kot, W. K.; Lappert, M. F.; Shalimoff, G. V.; Tian, S. J. Organomet. Chem. 2001, 636, 124.
- Evans, W. J.; Nyce, G. W.; Ziller, J. W. Organometallics 2001, 20, 5489.
- Evans, W. J.; Forrestal, K. J.; Leman, J. T.; Ziller, J. W. Organometallics 1996, 15, 527.
- Evans, W. J.; Seibel, C. A.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 6745.
- Evans, W. J.; Forrestal, K. J.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 9273.
- Evans, W. J. Adv. Organomet. Chem. 1985, 24, 131.
- Evans, W. J. Inorg. Chem. 2007, 46, 3435-3449.
- Evans, W. J. Polyhedron 1987, 6, 803.
- Evans, W. J.; DeCoster, D. M.; Greaves, J. Macromolecules. 1995, 28, 7929.
- Evans, W. J.; Ulibarri, T. A.; Chamberlain, L. R.; Ziller, J. W.; Alvarez, D. Organometallics 1990, 9, 2124.
- Evans, W. J.; Davis, B. L.; Perotti, J. M.; Kozimor, S.; Ziller, J. W. Manuscript in preparation.
- Evans, W. J.; Forrestal, K. J.; Ziller, J. W. J. Am. Chem. Soc. 1995, 117, 12635.
- Evans, W. J. Coord. Chem. Rev. 2000, 206, 263.
