Frustrated Lewis pair
In chemistry, a frustrated Lewis pair (FLP) is a compound or mixture containing a Lewis acid and a Lewis base that, because of steric hindrance, cannot combine to form a classical adduct.[1] Many kinds of FLPs have been devised, and many simple substrates exhibit activation.[2][3]
The discovery that some FLPs split H2[4] triggered a rapid growth of research into FLPs. Because of their "unquenched" reactivity, such systems are reactive toward substrates that can undergo heterolysis. For example, many FLPs split hydrogen molecules. Thus, a mixture of tricyclohexylphosphine (PCy3) and tris(pentafluorophenyl)borane reacts with hydrogen to give the respective phosphonium and borate ions:

This reactivity has been exploited to produce FLPs which catalyse hydrogenation reactions.[5]
Small molecule activation
Frustrated Lewis pairs have been shown to activate many small molecules, either by inducing heterolysis or by coordination.
Hydrogen
The discovery that some FLPs are able to split, and therefore activate, H2[4] triggered a rapid growth of research into this area. The activation and therefore use of H2 is important for many chemical and biological transformations. Using FLPs to liberate H2 is metal-free, this is beneficial due to the cost and limited supply of some transition metals commonly used to activate H2 (Ni, Pd, Pt).[6] FLP systems are reactive toward substrates that can undergo heterolysis (e.g hydrogen) due to the "unquenched" reactivity of such systems. For example, it has been previously shown that a mixture of tricyclohexylphosphine (PCy3) and tris(pentafluorophenyl)borane reacts with H2 to give the respective phosphonium and borate ions:
In this reaction, PCy3 (the Lewis Base) and B(C6F5)3 (the Lewis Acid) cannot form an adduct due to the steric hindrance from the bulky cyclohexyl and pentafluorophenyl groups. The proton on the phosphorus and hydride from the borate are now ‘activated’ and can subsequently be ‘delivered’ to an organic substrate, resulting in hydrogenation.
Mechanism of dihydrogen activation by FLP
The mechanism for the activation of H2 by FLPs has been discussed for both the intermolecular and intramolecular cases. Intermolecular FLPs are where the Lewis Base is a separate molecule to the Lewis Acid, it is thought that these individual molecules interact through secondary London dispersion interactions to bring the Lewis Base and Acid together (a pre-organisational effect) where small molecules may then interact with the FLPs. The experimental evidence for this type of interaction at the molecular level is unclear. However, there is supporting evidence for this type of interaction based on computational DFT (Density Functional Theory) studies. Intramolecular FLPs are where the Lewis Acid and Lewis Base are combined in one molecule by a covalent linker. Despite the improved ‘pre-organisational effects’, rigid intramolecular FLP frameworks are thought to have a reduced reactivity to small molecules due to a reduction in flexibility.
Dihydrogen activation example
The activation of H2 using FLPs was first reported in 2006.[4] In this report a phosphonium-borate species was observed to undergo the thermal loss of a H2 molecule, to generate the phosphine and the borane. The phosphonium-borate species 1 was prepared from B(C6F5)3 and (C6H2Me3)2PH, due to the steric demands of both of these molecules a traditional adduct could not be formed and the zwitterionic phosphonic-boronate was prepared [Figure 1]. A “H-for-F exchange” was carried out to give 2 which was stable to moisture and air. This salt will release molecular H2 when heated above 100 °C, a colour change was also observed from the colourless 2 to the orange-red 3 (both in THF). This reaction is reversed by reacting 3 with H2 at 25 ℃ where the colour change is reversed and 2 is reformed. The reaction of H2 with 3 was successful for temperatures as low as 25 ℃. Recrystallisation of 3 from THF afforded colourless crystals of the adduct 4.
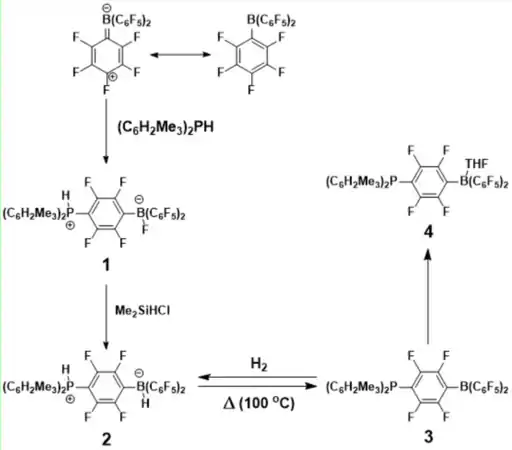
Other small molecule substrates
FLPs are also reactive toward many unsaturated substrates beyond H2. Some FLPs react with CO2, specifically in the deoxygenative reduction of CO2 to methane.[7]
Ethylene also reacts with FLPs:[8]

For acid-base pairs to behave both nucleophilically and electrophilically at the same time offers a method for the ring-opening of cyclic ethers such as THF, 2,5-dihydrofuran, coumaran, and dioxane.[9]
Use in catalysis
Imine, nitrile and aziridine hydrogenation

Reduction of imines, nitriles, and aziridines to primary and secondary amines traditionally is effected by metal hydride reagents, e.g. lithium aluminium hydride and sodium cyanoborohydride. Hydrogenations of these unsaturated substrates can be effected by metal-catalyzed reactions. Metal-free catalytic hydrogenation was carried out using the phosphonium borate catalyst (R2PH)(C6F4)BH(C6F5)2 (R = 2,4,6-Me3C6H2) 1. This type of metal-free hydrogenation has the potential to replace high cost metal catalyst.
The mechanism of imine reduction is proposed to involve protonation at nitrogen giving the iminium salt. The basicity of the nitrogen centre determines the rate of reaction. More electron rich imines reduce at faster rates than electron poor imines. The resulting iminium center undergoes nucleophilic attack by the borohydride anion to form the amine. Small amines bind to the borane, quenching further reactions. This problem can be overcome using various methods: 1) Application of elevated temperatures 2) Using sterically bulky imine substituents 3) Protecting the imine with the B(C6F5)3group, which also serves as a Lewis acid promoter.[10]
Enantioselective imine hydrogenation
Chiral boronate Lewis acid derived from (1R)-(+)-camphor form a frustrated Lewis pair with tBu3P, which is isolable as a salt. This FLP catalyses the enantioselective hydrogenation of some aryl imines in high yield but modest ee (up to 83%).
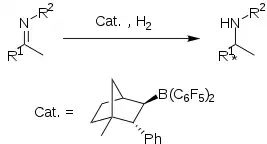
Although conceptually interesting, the protocol suffers from lack of generality. It was found that increasing steric bulk of the imine substituents lead to decreased yield and ee of the amine product. methoxy-substituted imines exhibit to superior yield and ee's.[10]
Asymmetric hydrosilylations
A group of catalysts, Frustrated Lewis pairs of chiral alkenylboranes and phosphines are beneficial for asymmetric Piers-type hydrosilylations of 1,2-dicarbonyl compounds and alpha-keto Esters, resulting in products of high yield enantioselectivity. However, in comparison conventional Piers-type hydrosilyation, asymmetric Piers-type hydrosilylations are not as well developed
In the following example, the chiral alkenylborane is formed in situ from chiral diyne and the HB(C6F5)2. Heterolytic cleavage of Si-H bond from PhMe2SiH by the FLP catalyst, forms a silylium and hydridoborate ionic complex.[11]
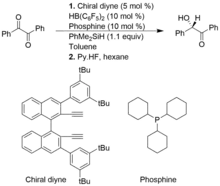
Alkyne hydrogenation
Metal free hydrogenation of unactivated internal alkynes to cis-alkenes is readily achieved using FLP-based catalysts.[12] The condition for this reaction were relatively mild utilising 2 bar of H2. In terms of mechanism, the alkyne material is first hydroborated and then the resulting vinylborane-based FLP can then activate dihydrogen. A protodeborylation step releases the cis-alkene product, which is obtained due to the syn-hydroborylation process, and regenerating the catalyst. While active for alkyne hydrogenation the FLP-based catalysts do not however facilitate the hydrogenation of alkenes to alkanes.
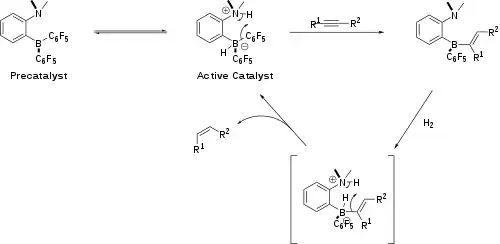
The reaction is a syn-hydroboration, and as a result a high cis selectivity is observed. At the final stage of the catalytic cycle the C6F5 group is cleaved more easily than an alkyl group, causing catalyst degradation rather than alkane release. The catalytic cycle has three steps:
- Substrate binding (the hydroboration of alkyne)
- H2 cleavage with vinylborane, followed by intramolecular protodeborylation of vinyl substituent, recovering N,N-Dimethyl-2-[(pentafluorophenyl)boryl]aniline
- Release of the cis-alkene
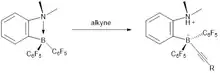
With internal alkynes, a competitive reaction occurs where the proton bound to the nitrogen can be added to the fluorobenzenes. Therefore, this addition does not proceed that much, the formation of the alkene seems favoured.
But terminal alkynes do not bind to the boron through hydroboration but rather through C-H activation. Thus, the addition of the proton to the alkyne will result in the initial terminal alkyne. Hence this hydrogenation process is not suitable to terminal alkynes and will only give pentafluorobenzene.
The metal free hydrogenation of terminal alkynes to the respective alkenes was recently achieved using a pyridone borane based system.[13] This system activates hydrogen readily at room temperature yielding a pyridone borane complex.[14] Dissociation of this complex allows hydroboration of an alkyne by the free borane. Upon protodeborylation by the free pyridone the cis alkene is generated. Hydrogenation of terminal alkynes is possible with this system, because the C-H activation is reversible and competes with hydrogen activation.
Borylation
Amine-borane FLPs catalyse the borylation of electron-rich aromatic heterocycles (Scheme 1).[15] The reaction is driven by release of hydrogen via C-H activation by the FLP. Aromatic borylations are often used in pharmaceutical development, particularly due to the abundance, low cost and low toxicity of boron compounds compared to noble metals.,
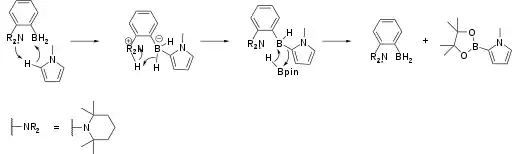
The substrate for the reaction has two main requirements, strongly linked to the mechanism of borylation. First, the substrate must be electron rich, exemplified by the absence of a reaction with thiophene, whereas its more electron rich derivatives - methoxythiophene and 3,4-ethylenedioxythiophene - can undergo a reaction with the amino-borane. Furthermore, substitution of 1-methylpyrrole (which can react) with the strongly electron withdrawing tertbutyloxycarbonyl (Boc) group at the 2-position completely inhibits the reaction. The second requirement is for the absence of basic amine groups in the substrate, which would otherwise form an unwanted adduct. This can be illustrated by the lack of a reaction with pyrrole, whereas both 1-methyl and N-benzylpyrrole derivatives are able to react.
Further work by the same authors revealed that simply piperidine as the amine R group (as opposed to tetramethylpiperidine, pictured above) accelerated the rate of reaction. Through kinetic and DFT studies the authors proposed that the C-H activation step was more facile than with larger substituents.[16]
Dearomatisation can also be achieved under similar conditions but using N-tosyl indoles. Syn-hyrdoborylated indolines are obtained.[17]
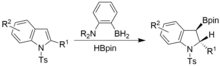
Borylation of S-H bonds in thiols by a dehydrogenative process has also been observed. Alcohols and amines such as tert-Butanol and tert-Butylamine form stable products that prevent catalysis due to a strong π-bond between the N/O atom’s lone pair and boron, whereas the same is not true for thiols, thus allowing for successful catalysis. In addition, successful borylation of Se-H bonds has been achieved. In all cases, the formation of H2 gas is a strong driving force for the reactions.[18]
Carbon capture
FLP chemistry is conceptually relevant to carbon capture.[19] Both an intermolecular (Scheme 1) and intramolecular (Scheme 2) FLP consisting of a phosphine and a borane were used to selectively capture and release carbon dioxide. When a solution of the FLP was covered by an atmosphere of CO2 at room temperature, the FLP-CO2 compound immediately precipitated as a white solid.[19][20]
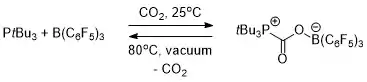
Heating the intermolecular FLP-CO2 compound in bromobenzene at 80 °C under vacuum for 5 hours caused the release of around half of the CO2 and regenerating the two constituent components of the FLP. After several more hours of sitting at room temperature under vacuum, total release of CO2 and FLP regeneration had occurred.[19]
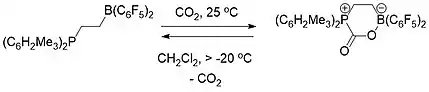
The intramolecular FLP-CO2 compound by contrast was stable as a solid at room temperature but fully decomposed at temperatures above -20 °C as a solution in dichloromethane releasing CO2 and regenerating the FLP molecule.[19]
This method of FLP carbon capture can be adapted to work in flow chemistry systems.[21]
References
- Stephan, Douglas W (2008). "Frustrated Lewis pairs: a concept for new reactivity and catalysis". Org. Biomol. Chem. 6 (9): 1535–1539. doi:10.1039/b802575b. PMID 18421382.

- Stephan, Douglas W.; Erker, Gerhard (2010). "Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More". Angewandte Chemie International Edition. 49 (1): 46–76. doi:10.1002/anie.200903708. ISSN 1433-7851. PMID 20025001.
- Stephan, Douglas W.; Erker, Gerhard (2017). "Frustrated Lewis pair chemistry". Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 375 (2101): 20170239. Bibcode:2017RSPTA.37570239S. doi:10.1098/rsta.2017.0239. ISSN 1364-503X. PMC 5540845. PMID 28739971.

- Welch, Gregory C.; Juan, Ronan R. San; Masuda, Jason D.; Stephan, Douglas W. (2006). "Reversible, Metal-Free Hydrogen Activation". Science. 314 (5802): 1124–1126. Bibcode:2006Sci...314.1124W. doi:10.1126/science.1134230. ISSN 0036-8075. PMID 17110572.
- Lam, Jolie; Szkop, Kevin M.; Mosaferi, Eliar; Stephan, Douglas W. (2018). "FLP catalysis: main group hydrogenations of organic unsaturated substrates". Chemical Society Reviews. 41 (13): 3592–3612. doi:10.1039/C8CS00277K. PMID 30178796.
- Welch, Gregory C.; Juan, Ronan R. San; Masuda, Jason D.; Stephan, Douglas W. (2006-11-17). "Reversible, Metal-Free Hydrogen Activation". Science. 314 (5802): 1124–1126. Bibcode:2006Sci...314.1124W. doi:10.1126/science.1134230. ISSN 0036-8075. PMID 17110572.
- Berkefeld, Andreas; Piers, Warren E.; Parvez, Masood (2010-08-11). "Tandem Frustrated Lewis Pair/Tris(pentafluorophenyl)borane-Catalyzed Deoxygenative Hydrosilylation of Carbon Dioxide". Journal of the American Chemical Society. 132 (31): 10660–10661. doi:10.1021/ja105320c. ISSN 0002-7863. PMID 20681691.
- Stephan, D. W. (2009). ""Frustrated Lewis Pairs": A New Strategy to Small Molecule Activation and Hydrogenation Catalysis". Dalton Trans (17): 3129–3136. doi:10.1039/b819621d. PMID 19421613.
- Tochertermam, W (1966). "Structures and Reactions of Organic ate-Complexes". Angew. Chem. Int. Ed. 5 (4): 351–171. doi:10.1002/anie.196603511.
- Chen, Dianjun; Wang, Yutian; Klankermayer, Jürgen (2010-12-03). "Enantioselective Hydrogenation with Chiral Frustrated Lewis Pairs". Angewandte Chemie International Edition. 49 (49): 9475–9478. doi:10.1002/anie.201004525. ISSN 1521-3773. PMID 21031385.
- Ren, Xiaoyu; Du, Haifeng (2016-01-15). "Chiral Frustrated Lewis Pairs Catalyzed Highly Enantioselective Hydrosilylations of 1,2-Dicarbonyl Compounds". Journal of the American Chemical Society. 138 (3): 810–813. doi:10.1021/jacs.5b13104. ISSN 0002-7863. PMID 26750998.
- Chernichenko, Konstantin; Madarász, Ádám; Pápai, Imre; Nieger, Martin; Leskelä, Markku; Repo, Timo (2013). "A frustrated-Lewis-pair approach to catalytic reduction of alkynes to cis-alkenes" (PDF). Nature Chemistry. 5 (8): 718–723. Bibcode:2013NatCh...5..718C. doi:10.1038/nchem.1693. PMID 23881505.
- Wech, Felix; Hasenbeck, Max; Gellrich, Urs (2020-09-18). "Semihydrogenation of Alkynes Catalyzed by a Pyridone Borane Complex: Frustrated Lewis Pair Reactivity and Boron–Ligand Cooperation in Concert". Chemistry – A European Journal: chem.202001276. doi:10.1002/chem.202001276. ISSN 0947-6539.
- Gellrich, Urs (2018). "Reversible Hydrogen Activation by a Pyridonate Borane Complex: Combining Frustrated Lewis Pair Reactivity with Boron-Ligand Cooperation". Angewandte Chemie International Edition. 57 (17): 4779–4782. doi:10.1002/anie.201713119. ISSN 1521-3773.
- Légaré, Marc A.; Courtmanche, Marc A.; Rochette, Étienne; Fontaine, Frédéric G. (2015-07-30). "Metal-free catalytic C-H bond activation and borylation of heteroarenes". Science. 349 (6247): 513–516. Bibcode:2015Sci...349..513L. doi:10.1126/science.aab3591. hdl:20.500.11794/30087. ISSN 0036-8075. PMID 26228143.
- Légaré Lavergne, Julien; Jayaraman, Arumugam; Misal Castro, Luis C.; Rochette, Étienne; Fontaine, Frédéric-Georges (2017-10-06). "Metal-Free Borylation of Heteroarenes Using Ambiphilic Aminoboranes: On the Importance of Sterics in Frustrated Lewis Pair C–H Bond Activation". Journal of the American Chemical Society. 139 (41): 14714–14723. doi:10.1021/jacs.7b08143. hdl:20.500.11794/30144. ISSN 0002-7863. PMID 28901757.
- Jayaraman, Arumugam; Misal Castro, Luis C.; Desrosiers, Vincent; Fontaine, Frédéric-Georges (2018). "Metal-free borylative dearomatization of indoles: exploring the divergent reactivity of aminoborane C–H borylation catalysts". Chemical Science. 9 (22): 5057–5063. doi:10.1039/c8sc01093e. ISSN 2041-6520. PMC 5994747. PMID 29938036.
- Rochette, Étienne; Boutin, Hugo; Fontaine, Frédéric-Georges (2017-06-30). "Frustrated Lewis Pair Catalyzed S–H Bond Borylation". Organometallics. 36 (15): 2870–2876. doi:10.1021/acs.organomet.7b00346. hdl:20.500.11794/30088. ISSN 0276-7333.
- Mömming, Cornelia M.; Otten, Edwin; Kehr, Gerald; Fröhlich, Roland; Grimme, Stefan; Stephan, Douglas W.; Erker, Gerhard (2009-08-24). "Reversible Metal-Free Carbon Dioxide Binding by Frustrated Lewis Pairs" (PDF). Angewandte Chemie International Edition. 48 (36): 6643–6646. doi:10.1002/anie.200901636. ISSN 1433-7851. PMID 19569151.
- Stephan, Douglas W.; Erker, Gerhard (2015-05-14). "Frustrated Lewis Pair Chemistry: Development and Perspectives". Angewandte Chemie International Edition. 54 (22): 6400–6441. doi:10.1002/anie.201409800. ISSN 1433-7851. PMID 25974714.
- Abolhasani, Milad; Günther, Axel; Kumacheva, Eugenia (2014-06-24). "Microfluidic Studies of Carbon Dioxide". Angewandte Chemie International Edition. 53 (31): 7992–8002. doi:10.1002/anie.201403719. ISSN 1433-7851. PMID 24961230.