H3R17me2
H3R17me2 is an epigenetic modification to the DNA packaging protein histone H3. It is a mark that indicates the di-methylation at the 17th arginine residue of the histone H3 protein. In epigenetics, arginine methylation of histones H3 and H4 is associated with a more accessible chromatin structure and thus higher levels of transcription. The existence of arginine demethylases that could reverse arginine methylation is controversial.[1]
Nomenclature
The name of this modification indicates dimethylation of arginine 17 on histone H3 protein subunit: [2]
| Abbr. | Meaning |
| H3 | H3 family of histones |
| R | standard abbreviation for arginine |
| 17 | position of amino acid residue
(counting from N-terminus) |
| me | methyl group |
| 2 | number of methyl groups added |
Arginine
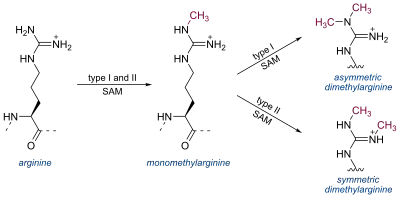
Arginine can be methylated once (monomethylated arginine) or twice (dimethylated arginine). Methylation of arginine residues is catalyzed by three different classes of protein arginine methyltransferases.[1]
Arginine methylation affects the interactions between proteins and has been implicated in a variety of cellular processes, including protein trafficking, signal transduction, and transcriptional regulation.[3]
Arginine methylation plays a major role in gene regulation because of the ability of the PRMTs to deposit key activating (histone H4R3me2a, H3R2me2s, H3R17me2a, H3R26me2a) or repressive (H3R2me2a, H3R8me2a, H3R8me2s, H4R3me2s) histone marks.
Histone modifications
THE genomic DNA of eukaryotic cells is wrapped around special protein molecules known as histones. The complexes formed by the looping of the DNA are known as chromatin.
Mechanism and function of modification
Methylation of H3R17 is mediated by CARM1 and is recruited to promoter upon gene activation along with acetyltransferases and activates transcription. When CARM1 is recruited to transcriptional promoters the histone H3 is methylated (H3R17me2 & H3R26me2). H3R17 dimethylation as a critical epigenetic mark in starvation-induced autophagy[4]
Epigenetic implications
The post-translational modification of histone tails by either histone-modifying complexes or chromatin remodeling complexes is interpreted by the cell and leads to complex, combinatorial transcriptional output. It is thought that a histone code dictates the expression of genes by a complex interaction between the histones in a particular region.[5] The current understanding and interpretation of histones comes from two large scale projects: ENCODE and the Epigenomic roadmap.[6] The purpose of the epigenomic study was to investigate epigenetic changes across the entire genome. This led to chromatin states, which define genomic regions by grouping different proteins and/or histone modifications together. Chromatin states were investigated in Drosophila cells by looking at the binding location of proteins in the genome. Use of ChIP-sequencing revealed regions in the genome characterized by different banding.[7] Different developmental stages were profiled in Drosophila as well, an emphasis was placed on histone modification relevance.[8] A look in to the data obtained led to the definition of chromatin states based on histone modifications.[9] Certain modifications were mapped and enrichment was seen to localize in certain genomic regions.
The human genome is annotated with chromatin states. These annotated states can be used as new ways to annotate a genome independently of the underlying genome sequence. This independence from the DNA sequence enforces the epigenetic nature of histone modifications. Chromatin states are also useful in identifying regulatory elements that have no defined sequence, such as enhancers. This additional level of annotation allows for a deeper understanding of cell specific gene regulation.[10]
Clinical significance
CARM1 knockout mice are smaller and die shortly after birth. CARM1 is required for the epigenetic maintenance of pluripotency and self-renewal, as it methylates H3R17 and H3R26 at core pluripotency genes such as Oct4, Sox2, and Nanog.[1]
CARM1-mediated methylation of H3R17 is needed to establish and maintain the astroglial lineage. The absence of H3R17me2a downregulates miR-92a levels, and this miRNA is known to participate in neural development.[1]
CARM1 was indirectly associated with diabetic retinopathy progression, as it and its associated H3R17me2a mark increased with high glucose, promoting apoptosis of the retinal pigment epithelial cells.[1]
Methods
The histone mark H3K4me1 can be detected in a variety of ways:
1. Chromatin Immunoprecipitation Sequencing (ChIP-sequencing) measures the amount of DNA enrichment once bound to a targeted protein and immunoprecipitated. It results in good optimization and is used in vivo to reveal DNA-protein binding occurring in cells. ChIP-Seq can be used to identify and quantify various DNA fragments for different histone modifications along a genomic region.[11]
2. Micrococcal Nuclease sequencing (MNase-seq) is used to investigate regions that are bound by well-positioned nucleosomes. Use of the micrococcal nuclease enzyme is employed to identify nucleosome positioning. Well-positioned nucleosomes are seen to have enrichment of sequences.[12]
3. Assay for transposase accessible chromatin sequencing (ATAC-seq) is used to look in to regions that are nucleosome free (open chromatin). It uses hyperactive Tn5 transposon to highlight nucleosome localisation.[13][14][15]
References
- Blanc, Roméo S.; Richard, Stéphane (2017). "Arginine Methylation: The Coming of Age". Molecular Cell. 65 (1): 8–24. doi:10.1016/j.molcel.2016.11.003. PMID 28061334.
- Huang, Suming; Litt, Michael D.; Ann Blakey, C. (30 November 2015). Epigenetic Gene Expression and Regulation. pp. 21–38. ISBN 9780127999586.
- McBride, A.; Silver, P. (2001). "State of the Arg: Protein Methylation at Arginine Comes of Age". Cell. 106 (1): 5–8. doi:10.1016/S0092-8674(01)00423-8. PMID 11461695. S2CID 17755108.
- Shin, Hi-Jai R.; Kim, Hyunkyung; Kim, Keun Il; Baek, Sung Hee (2016). "Epigenetic and transcriptional regulation of autophagy". Autophagy. 12 (11): 2248–2249. doi:10.1080/15548627.2016.1214780. PMC 5103355. PMID 27487449.
- Jenuwein T, Allis CD (August 2001). "Translating the histone code". Science. 293 (5532): 1074–80. doi:10.1126/science.1063127. PMID 11498575. S2CID 1883924.
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, et al. (The ENCODE Project Consortium) (June 2007). "Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project". Nature. 447 (7146): 799–816. Bibcode:2007Natur.447..799B. doi:10.1038/nature05874. PMC 2212820. PMID 17571346.
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B (October 2010). "Systematic protein location mapping reveals five principal chromatin types in Drosophila cells". Cell. 143 (2): 212–24. doi:10.1016/j.cell.2010.09.009. PMC 3119929. PMID 20888037.
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, et al. (modENCODE Consortium) (December 2010). "Identification of functional elements and regulatory circuits by Drosophila modENCODE". Science. 330 (6012): 1787–97. Bibcode:2010Sci...330.1787R. doi:10.1126/science.1198374. PMC 3192495. PMID 21177974.
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, et al. (March 2011). "Comprehensive analysis of the chromatin landscape in Drosophila melanogaster". Nature. 471 (7339): 480–5. Bibcode:2011Natur.471..480K. doi:10.1038/nature09725. PMC 3109908. PMID 21179089.
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, et al. (Roadmap Epigenomics Consortium) (February 2015). "Integrative analysis of 111 reference human epigenomes". Nature. 518 (7539): 317–30. Bibcode:2015Natur.518..317.. doi:10.1038/nature14248. PMC 4530010. PMID 25693563.
- "Whole-Genome Chromatin IP Sequencing (ChIP-Seq)" (PDF). Illumina. Retrieved 23 October 2019.
- "MAINE-Seq/Mnase-Seq". illumina. Retrieved 23 October 2019.
- Buenrostro, Jason D.; Wu, Beijing; Chang, Howard Y.; Greenleaf, William J. (2015). "ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide". Current Protocols in Molecular Biology. 109: 21.29.1–21.29.9. doi:10.1002/0471142727.mb2129s109. ISBN 9780471142720. PMC 4374986. PMID 25559105.
- Schep, Alicia N.; Buenrostro, Jason D.; Denny, Sarah K.; Schwartz, Katja; Sherlock, Gavin; Greenleaf, William J. (2015). "Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions". Genome Research. 25 (11): 1757–1770. doi:10.1101/gr.192294.115. ISSN 1088-9051. PMC 4617971. PMID 26314830.
- Song, L.; Crawford, G. E. (2010). "DNase-seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells". Cold Spring Harbor Protocols. 2010 (2): pdb.prot5384. doi:10.1101/pdb.prot5384. ISSN 1559-6095. PMC 3627383. PMID 20150147.