Hydroboration–oxidation reaction
In organic chemistry, the hydroboration–oxidation reaction is a two-step hydration reaction that converts an alkene into an alcohol.[1] The process results in the syn addition of a hydrogen and a hydroxyl group where the double bond had been. Hydroboration–oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction thus provides a more stereospecific and complementary regiochemical alternative to other hydration reactions such as acid-catalyzed addition and the oxymercuration–reduction process. The reaction was first reported by Herbert C. Brown in the late 1950s[2] and it was recognized in his receiving the Nobel Prize in Chemistry in 1979.
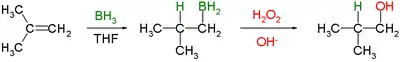
The general form of the reaction is as follows:
Tetrahydrofuran (THF) is the archetypal solvent used for hydroboration.
Mechanism and scope
Hydroboration step
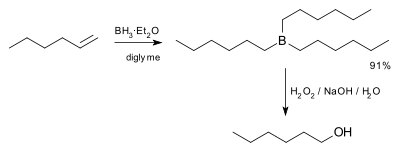
In the first step, borane (BH3) adds to the double bond, transferring one of the hydrogen atoms to the carbon adjacent to the one that becomes bonded to the boron. This hydroboration is repeated two additional times, successively reacting each B–H bond so that three alkenes add to each BH3. The resulting trialkylborane is treated with hydrogen peroxide in the second step. This process replaces the B-C bonds with HO-C bonds. The boron reagent is converted to boric acid. The reaction was originally described by H.C. Brown in 1957 for the conversion of 1-hexene into 1-hexanol.[3]
Knowing that the group containing the boron will be replaced by a hydroxyl group, it can be seen that the initial hydroboration step determines the regioselectivity. Hydroboration proceeds in an antimarkovnikov manner. The reaction sequence is also stereospecific, giving syn addition (on the same face of the alkene): the hydroboration is syn-selective and the oxidation replaces the boron with hydroxyl having the same geometric position. Thus 1-methylcyclopentene reacts with diborane predominantly to give trans-1-hydroxy-2-methylcyclopentane[4]—the newly added H and OH are cis to each other.
Until all hydrogens attached to boron have been transferred away, the boron group BH2 will continue adding to more alkenes. This means that one mole of hydroborane will undergo the reaction with three moles of alkene. Furthermore, it is not necessary for the hydroborane to have more than one hydrogen. For example, reagents of the type R2BH are commonly used, where R can represents the remainder of the molecule. Such modified hydroboration reagents include 9-BBN, catecholborane, and disiamylborane.
Oxidation step
In the second step of the reaction sequence, the nucleophilic hydroperoxide anion attacks the boron atom. Alkyl migration to oxygen gives the alkyl borane with retention of stereochemistry (in reality, the reaction occurs via the trialkyl borate B(OR)3, rather than the monoalkyl borinic ester BH2OR).

The 'H' atom in the reaction comes from B2H6, the 'O' atom comes from hydrogen peroxide (H2O2) whereas the O attached 'H' atom comes from the solvent (refer mechanism).
Alkyne hydroboration
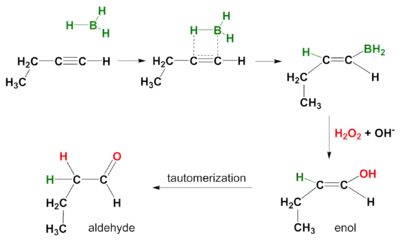
A hydroboration reaction also takes place on alkynes. Again the mode of action is syn and secondary reaction products are aldehydes from terminal alkynes and ketones from internal alkynes. In order to prevent hydroboration across both the pi-bonds, a bulky borane like disiamyl (di-sec-iso-amyl) borane is used.[5]
Alternative oxidations
Use of other oxidants instead of hydrogen peroxide can lead to carbonyl products rather than alcohols from alkenes. N-Methylmorpholine N-oxide with catalytic tetrapropylammonium perruthenate converts the alkylborane into a carbonyl, thus a ketone or aldehyde product depending on what other groups were attached to that carbon in the original alkene.[6] Various dichromates or related chromium(VI) reagents give ketones as well, but give carboxylic acids instead of aldehydes for terminal alkenes.[7]
References
- Loudon, Marc G. (2002). "Addition Reactions of Alkenes.". Organic Chemistry (Fourth ed.). New York: Oxford University Press. pp. 168–172. ISBN 0-19-511999-1.
- Brown, H. C.; Zweifel, G. (1959). "A STEREOSPECIFIC CIS HYDRATION OF THE DOUBLE BOND IN CYCLIC DERIVATIVES". Journal of the American Chemical Society. 81: 247. doi:10.1021/ja01510a059.
- Brown, H.; Rao, B. C. (1957). "Communications – Selective Conversion of Olefins into Organoboranes Through Competitive Hydroboration, Isomerization and Displacement Reactions". Journal of Organic Chemistry. 22 (9): 1137. doi:10.1021/jo01360a626.
- Hawthorne, M. F. (1961). "Amine Boranes. VIII. The Hydroboration of Terminal Olefins, Dienes and Terminal Acetylenes with Trimethylamine t-Butylborane". Journal of the American Chemical Society. 83 (11): 2541–2544. doi:10.1021/ja01472a027.
- Brown, H. C.; Gupta, S. K. (1972). "Catecholborane (1,3,2-benzodioxaorole) as a new, general monohydroboration reagent for alkynes. Convenient synthesis of alkeneboronic esters and acids from alkynes via hydroboration". Journal of the American Chemical Society. 94 (12): 4370. doi:10.1021/ja00767a072.
- Yates, Matthew H. (1997). "One-pot conversion of olefins to carbonyl compounds by hydroboration / NMO–TPAP oxidation". Tetrahedron Lett. 38: 2813–2816. doi:10.1016/S0040-4039(97)00476-0.
- Brown, Herbert C.; Kulkarni, Shekhar V.; Khanna, Vijay V.; Patil, Virendra D.; Racherla, Uday S. (1992). "Organoboranes for Synthesis. 14. Convenient Procedures for the Direct Oxidation of Organoboranes from Terminal Alkenes to Carboxylic Acids". J. Org. Chem. 57 (23): 6173–6177. doi:10.1021/jo00049a024.
External links
- Organic Chemistry Portal. Hydroboration (including recent literature). https://www.organic-chemistry.org/namedreactions/brown-hydroboration.shtm