Interatomic potential
Interatomic potentials are mathematical functions to calculate the potential energy of a system of atoms with given positions in space.[1][2][3][4] Interatomic potentials are widely used as the physical basis of molecular mechanics and molecular dynamics simulations in computational chemistry, computational physics and computational materials science to explain and predict materials properties. Examples of quantitative properties and qualitative phenomena that are explored with interatomic potentials include lattice parameters, surface energies, interfacial energies, adsorption, cohesion, thermal expansion, and elastic and plastic material behavior, as well as chemical reactions.[5][6][7][8][9][10][11]
Functional form
Interatomic potentials can be written as a series expansion of functional terms that depend on the position of one, two, three, etc. atoms at a time. Then the total potential of the system can be written as [3]


Here is the one-body term, the two-body term, the three body term, the number of atoms in the system, the position of atom , etc. , and are indices that loop over atom positions.








Note that in case the pair potential is given per atom pair, in the two-body term the potential should be multiplied by 1/2 as otherwise each bond is counted twice, and similarly the three-body term by 1/6.[3] Alternatively, the summation of the pair term can be restricted to cases and similarly for the three-body term , if the potential form is such that it is symmetric with respect to exchange of the and indices (this may not be the case for potentials for multielemental systems).


The one-body term is only meaningful if the atoms are in an external field (e.g. an electric field). In the absence of external fields, the potential should not depend on the absolute position of atoms, but only on the relative positions. This means that the functional form can be rewritten as a function of interatomic distances and angles between the bonds (vectors to neighbours) . Then, in the absence of external forces, the general form becomes




In the three-body term the interatomic distance is not needed since the three terms are sufficient to give the relative positions of three atoms in three-dimensional space. Any terms of order higher than 2 are also called many-body potentials. In some interatomic potentials the many-body interactions are embedded into the terms of a pair potential (see discussion on EAM-like and bond order potentials below).



In principle the sums in the expressions run over all atoms. However, if the range of the interatomic potential is finite, i.e. the potentials above some cutoff distance , the summing can be restricted to atoms within the cutoff distance of each other. By also using a cellular method for finding the neighbours,[1] the MD algorithm can be an O(N) algorithm. Potentials with an infinite range can be summed up efficiently by Ewald summation and its further developments.



Force calculation
The forces acting between atoms can be obtained by differentiation of the total energy with respect to atom positions. That is, to get the force on atom one should take the three-dimensional derivative (gradient) with respect to the position of atom :

For two-body potentials this gradient reduces, thanks to the symmetry with respect to in the potential form, to straightforward differentiation with respect to the interatomic distances . However, for many-body potentials (three-body, four-body, etc.) the differentiation becomes considerably more complex [12] [13] since the potential may not be any longer symmetric with respect to exchange. In other words, also the energy of atoms that are not direct neighbours of can depend on the position because of angular and other many-body terms, and hence contribute to the gradient .




Classes of interatomic potentials
Interatomic potentials come in many different varieties, with different physical motivations. Even for single well-known elements such as silicon, a wide variety of potentials quite different in functional form and motivation have been developed.[14] The true interatomic interactions are quantum mechanical in nature, and there is no known way in which the true interactions described by the Schrödinger equation or Dirac equation for all electrons and nuclei could be cast into an analytical functional form. Hence all analytical interatomic potentials are by necessity approximations.
Over time interatomic potentials have largely grown more complex and more accurate, although this is not strictly true.[15] This has included both increased descriptions of physics, as well as added parameters. Until recently, all interatomic potentials could be described as "parametric", having been developed and optimized with a fixed number of (physical) terms and parameters. New research focuses instead on non-parametric potentials which can be systematically improvable by using complex local atomic neighbor descriptors and separate mappings to predict system properties, such that the total number of terms and parameters are flexible. [16] These non-parameteric models can be significantly more accurate, but since they are not tied to physical forms and parameters, there are many potential issues surrounding extrapolation and uncertainties.
Pair potentials
The arguably simplest widely used interatomic interaction model is the Lennard-Jones potential [17]

where is the depth of the potential well and is the distance at which the potential crosses zero. The attractive term proportional to in the potential comes from the scaling of van der Waals forces, while the repulsive term is much more approximate (conveniently the square of the attractive term).[6] On its own, this potential is quantitatively accurate only for noble gases, but is also widely used for qualitative studies and in systems where dipole interactions are significant, particularly in chemistry force fields to describe intermolecular interactions.




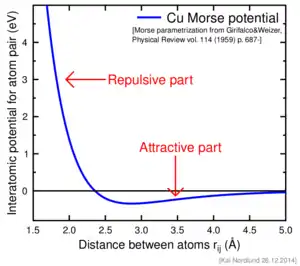
Another simple and widely used pair potential is the Morse potential, which consists simply of a sum of two exponentials.

Here is the equilibrium bond energy and the bond distance. The Morse potential has been applied to studies of molecular vibrations and solids ,[18] and also inspired the functional form of more accurate potentials such as the bond-order potentials.


Ionic materials are often described by a sum of a short-range repulsive term, such as the Buckingham pair potential, and a long-range Coulomb potential giving the ionic interactions between the ions forming the material. The short-range term for ionic materials can also be of many-body character .[19]
Pair potentials have some inherent limitations, such as the inability to describe all 3 elastic constants of cubic metals or correctly describe both cohesive energy and vacancy formation energy.[7] Therefore quantitative molecular dynamics simulations are carried out with various of many-body potentials.
Repulsive potentials
For very short interatomic separations, important in radiation material science, the interactions can be described quite accurately with screened Coulomb potentials which have the general form

Here, when . and are the charges of the interacting nuclei, and is the so-called screening parameter. A widely used popular screening function is the "Universal ZBL" one.[20] and more accurate ones can be obtained from all-electron quantum chemistry calculations [21] In binary collision approximation simulations this kind of potential can be used to describe the nuclear stopping power.





Many-body potentials
The Stillinger-Weber potential[22] is a potential that has a two-body and three-body terms of the standard form

where the three-body term describes how the potential energy changes with bond bending. It was originally developed for pure Si, but has been extended to many other elements and compounds [23] [24] and also formed the basis for other Si potentials.[25] [26]
Metals are very commonly described with what can be called "EAM-like" potentials, i.e. potentials that share the same functional form as the embedded atom model. In these potentials, the total potential energy is written

where is a so-called embedding function (not to be confused with the force ) that is a function of the sum of the so-called electron density . is a pair potential that usually is purely repulsive. In the original formulation [27][28] the electron density function was obtained from true atomic electron densities, and the embedding function was motivated from density-functional theory as the energy needed to 'embed' an atom into the electron density. .[29] However, many other potentials used for metals share the same functional form but motivate the terms differently, e.g. based on tight-binding theory [30][31] [32] or other motivations [33] [34] .[35]



EAM-like potentials are usually implemented as numerical tables. A collection of tables is available at the interatomic potential repository at NIST
Covalently bonded materials are often described by bond order potentials, sometimes also called Tersoff-like or Brenner-like potentials. [10] [36] [37]
These have in general a form that resembles a pair potential:

where the repulsive and attractive part are simple exponential functions similar to those in the Morse potential. However, the strength is modified by the environment of the atom via the term. If implemented without an explicit angular dependence, these potentials can be shown to be mathematically equivalent to some varieties of EAM-like potentials [38] [39] Thanks to this equivalence, the bond-order potential formalism has been implemented also for many metal-covalent mixed materials.[39][40] [41] [42]

EAM potentials have also been extended to describe covalent bonding by adding angular-dependent terms to the electron density function , in what is called the modified embedded atom method (MEAM).[43][44][45]

Force fields
A force field is the collection of parameters to describe the physical interactions between atoms or physical units (up to ~108) using a given energy expression. The term force field characterizes the collection of parameters for a given interatomic potential (energy function) and is often used within the computational chemistry community.[46] The force field makes the difference between good and poor models. Force fields are used for the simulation of metals, ceramics, molecules, chemistry, and biological systems, covering the entire periodic table and multiphase materials. Today's performance is among the best for solid-state materials[47][48] and for biomacromolecules,[49] whereby biomacromolecules were the primary focus of force fields from the 1970s to the early 2000s. Force fields range from relatively simple and interpretable fixed-bond models (e.g. Interface force field,[46] CHARMM,[50] and COMPASS) to explicitly reactive models with many adjustable fit parameters (e.g. ReaxFF) and machine learning models.
Non-parametric potentials
It should first be noted that non-parametric potentials are often referred to as "machine learning" potentials. While the descriptor/mapping forms of non-parametric models are closely related to machine learning in general and their complex nature make machine learning fitting optimizations almost necessary, differentiation is important in that parametric models can also be optimized using machine learning.
Current research in interatomic potentials involves using systematically improvable, non-parameteric mathematical forms and increasingly complex machine learning methods. The total energy is then written

where is a mathematical representation of the atomic environment surrounding the atom , known as the descriptor.[51] is a machine-learning model that provides a prediction for the energy of atom based on the descriptor output. An accurate machine-learning potential requires both a robust descriptor and a suitable machine learning framework. The simplest descriptor is the set of interatomic distances from atom to its neighbours, yielding a machine-learned pair potential. However, more complex many-body descriptors are needed to produce highly accurate potentials. [51] It is also possible to use a linear combination of multiple descriptors with associated machine-learning models.[52] Potentials have been constructed using a variety of machine-learning methods, descriptors, and mappings, including neural networks,[53] Gaussian process regression,[54][55] and linear regression.[56][16]


A non-parametric potential is most often trained to total energies, forces, and/or stresses obtained from quantum-level calculations, such as density functional theory, as with most modern potentials. However, the accuracy of a machine-learning potential can be converged to be comparable with the underlying quantum calculations, unlike analytical models. Hence, they are in general more accurate than traditional analytical potentials, but they are correspondingly less able to extrapolate. Further, owing to the complexity of the machine-learning model and the descriptors, they are computationally far more expensive than their analytical counterparts.
Non-parametric, machine learned potentials may also be combined with parametric, analytical potentials, for example to include known physics such as the screened Coulomb repulsion,[57] or to impose physical constraints on the predictions.[58]
Potential fitting
Since the interatomic potentials are approximations, they by necessity all involve parameters that need to be adjusted to some reference values. In simple potentials such as the Lennard-Jones and Morse ones, the parameters are interpretable and can be set to match e.g. the equilibrium bond length and bond strength of a dimer molecule or the surface energy of a solid .[59][60] Lennard-Jones potential can typically describe the lattice parameters, surface energies, and approximate mechanical properties.[61] Many-body potentials often contain tens or even hundreds of adjustable parameters with limited interpretability and no compatibility with common interatomic potentials for bonded molecules. Such parameter sets can be fit to a larger set of experimental data, or materials properties derived from less reliable data such as from density-functional theory.[62][63] For solids, a many-body potential can often describe the lattice constant of the equilibrium crystal structure, the cohesive energy, and linear elastic constants, as well as basic point defect properties of all the elements and stable compounds well, although deviations in surface energies often exceed 50%. [26][39][41][42][61][46] [64] [65] [66] Non-parameteric potentials in turn contain hundreds or even thousands of independent parameters to fit. For any but the simplest model forms, sophisticated optimization and machine learning methods are necessary for useful potentials.
The aim of most potential functions and fitting is to make the potential transferable, i.e. that it can describe materials properties that are clearly different from those it was fitted to (for examples of potentials explicitly aiming for this, see e.g.[67][68][69][70][71]). Key aspects here are the correct representation of chemical bonding, validation of structures and energies, as well as interpretability of all parameters.[47] Full transferability and interpretability is reached with the Interface force field (IFF).[46] An example of partial transferability, a review of interatomic potentials of Si describes that Stillinger-Weber and Tersoff III potentials for Si can describe several (but not all) materials properties they were not fitted to.[14]
The NIST interatomic potential repository provides a collection of fitted interatomic potentials, either as fitted parameter values or numerical tables of the potential functions.[72] The OpenKIM [73] project also provides a repository of fitted potentials, along with collections of validation tests and a software framework for promoting reproducibility in molecular simulations using interatomic potentials.
Reliability of interatomic potentials
Classical interatomic potentials often exceed the accuracy of simplified quantum mechanical methods such as density functional theory at a million times lower computational cost.[47] The use of interatomic potentials is recommended for the simulation of nanomaterials, biomacromolecules, and electrolytes from atoms up to millions of atoms at the 100 nm scale and beyond. As a limitation, electron densities and quantum processes at the local scale of hundreds of atoms are not included. When of interest, higher level quantum chemistry methods can be locally used.[74]
The robustness of a model at different conditions other than those used in the fitting process is often measured in terms of transferability of the potential.
See also
References
- M. P. Allen and D. J. Tildesley. Computer Simulation of Liquids. Oxford University Press, Oxford, England, 1989.
- Daan Frenkel and Berend Smit. Understanding molecular simulation: from algorithms to applications. Academic Press, San Diego, second edition, 2002.
- R. Lesar. Introduction to Computational Materials Science. Cambridge University Press, 2013.
- Brenner, D.W. (2000). "The Art and Science of an Analytic Potential". Physica Status Solidi B. 217 (1): 23–40. Bibcode:2000PSSBR.217...23B. doi:10.1002/(SICI)1521-3951(200001)217:1<23::AID-PSSB23>3.0.CO;2-N. ISSN 0370-1972.
- N. W. Ashcroft and N. D. Mermin. Solid State Physics.Saunders College, Philadelphia, 1976.
- Charles Kittel. Introduction to Solid State Physics. John Wiley & Sons, New York, third edition, 1968.
- Daw, Murray S.; Foiles, Stephen M.; Baskes, Michael I. (1993). "The embedded-atom method: a review of theory and applications". Materials Science Reports. 9 (7–8): 251–310. doi:10.1016/0920-2307(93)90001-U. ISSN 0920-2307.
- Tersoff J (April 1988). "New empirical approach for the structure and energy of covalent systems". Physical Review B. 37 (12): 6991–7000. Bibcode:1988PhRvB..37.6991T. doi:10.1103/physrevb.37.6991. PMID 9943969.
- FINNIS, M (2007). "Bond-order potentials through the ages". Progress in Materials Science. 52 (2–3): 133–153. doi:10.1016/j.pmatsci.2006.10.003. ISSN 0079-6425.
- Sinnott, Susan B.; Brenner, Donald W. (2012). "Three decades of many-body potentials in materials research". MRS Bulletin. 37 (5): 469–473. doi:10.1557/mrs.2012.88. ISSN 0883-7694.
- Bedford NM, Ramezani-Dakhel H, Slocik JM, Briggs BD, Ren Y, Frenkel AI, et al. (May 2015). "Elucidation of peptide-directed palladium surface structure for biologically tunable nanocatalysts". ACS Nano. 9 (5): 5082–92. doi:10.1021/acsnano.5b00168. PMID 25905675.
- Beardmore, Keith M.; Grønbech-Jensen, Niels (1 October 1999). "Direct simulation of ion-beam-induced stressing and amorphization of silicon". Physical Review B. 60 (18): 12610–12616. arXiv:cond-mat/9901319v2. Bibcode:1999PhRvB..6012610B. doi:10.1103/physrevb.60.12610. ISSN 0163-1829.
- Albe, Karsten; Nord, J.; Nordlund, K. (2009). "Dynamic charge-transfer bond-order potential for gallium nitride". Philosophical Magazine. 89 (34–36): 3477–3497. Bibcode:2009PMag...89.3477A. doi:10.1080/14786430903313708. ISSN 1478-6435.
- Balamane H, Halicioglu T, Tiller WA (July 1992). "Comparative study of silicon empirical interatomic potentials". Physical Review B. 46 (4): 2250–2279. Bibcode:1992PhRvB..46.2250B. doi:10.1103/physrevb.46.2250. PMID 10003901.
- Plimpton SJ, Thompson AP (2012). "Computational aspects of many-body potentials". MRS Bull. 37: 513–521.
- Shapeev, Alexander V. (2016-09-13). "Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials". Multiscale Modeling & Simulation. 14 (3): 1153–1173. arXiv:1512.06054. doi:10.1137/15M1054183. ISSN 1540-3459. S2CID 28970251.
- Lennard-Jones, J. E. (1924). "On the Determination of Molecular Fields". Proc. R. Soc. Lond. A. 106 (738): 463–477. Bibcode:1924RSPSA.106..463J. doi:10.1098/rspa.1924.0082..
- Girifalco, L. A.; Weizer, V. G. (1 April 1959). "Application of the Morse Potential Function to Cubic Metals". Physical Review. 114 (3): 687–690. Bibcode:1959PhRv..114..687G. doi:10.1103/physrev.114.687. hdl:10338.dmlcz/103074. ISSN 0031-899X.
- Feuston, B. P.; Garofalini, S. H. (1988). "Empirical three‐body potential for vitreous silica". The Journal of Chemical Physics. 89 (9): 5818–5824. Bibcode:1988JChPh..89.5818F. doi:10.1063/1.455531. ISSN 0021-9606.
- J. F. Ziegler, J. P. Biersack, and U. Littmark. The Stopping and Range of Ions in Matter. Pergamon, New York, 1985.
- Nordlund, K.; Runeberg, N.; Sundholm, D. (1997). "Repulsive interatomic potentials calculated using Hartree-Fock and density-functional theory methods". Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms. 132 (1): 45–54. Bibcode:1997NIMPB.132...45N. doi:10.1016/s0168-583x(97)00447-3. ISSN 0168-583X.
- Stillinger FH, Weber TA (April 1985). "Computer simulation of local order in condensed phases of silicon". Physical Review B. 31 (8): 5262–5271. Bibcode:1985PhRvB..31.5262S. doi:10.1103/physrevb.31.5262. PMID 9936488.
- Ichimura, M. (16 February 1996). "Stillinger-Weber potentials for III–V compound semiconductors and their application to the critical thickness calculation for InAs/GaAs". Physica Status Solidi A. 153 (2): 431–437. Bibcode:1996PSSAR.153..431I. doi:10.1002/pssa.2211530217. ISSN 0031-8965.
- Ohta, H.; Hamaguchi, S. (2001). "Classical interatomic potentials for si-o-f and si-o-cl systems". Journal of Chemical Physics. 115 (14): 6679–90. doi:10.1063/1.1400789. hdl:2433/50272.
- Bazant, M. Z.; Kaxiras, E.; Justo, J. F. (1997). "Environment-dependent interatomic potential for bulk silicon". Phys. Rev. B. 56 (14): 8542. arXiv:cond-mat/9704137. Bibcode:1997PhRvB..56.8542B. doi:10.1103/PhysRevB.56.8542. S2CID 17860100.
- Justo, João F.; Bazant, Martin Z.; Kaxiras, Efthimios; Bulatov, V. V.; Yip, Sidney (1 July 1998). "Interatomic potential for silicon defects and disordered phases". Physical Review B. 58 (5): 2539–2550. arXiv:cond-mat/9712058. Bibcode:1998PhRvB..58.2539J. doi:10.1103/physrevb.58.2539. ISSN 0163-1829.
- Foiles SM, Baskes MI, Daw MS (June 1986). "Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys". Physical Review B. 33 (12): 7983–7991. Bibcode:1986PhRvB..33.7983F. doi:10.1103/physrevb.33.7983. PMID 9938188.
- Foiles, S. M.; Baskes, M. I.; Daw, M. S. (15 June 1988). "Erratum: Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys". Physical Review B. 37 (17): 10378. doi:10.1103/physrevb.37.10378. ISSN 0163-1829.
- Puska, M. J.; Nieminen, R. M.; Manninen, M. (15 September 1981). "Atoms embedded in an electron gas: Immersion energies". Physical Review B. 24 (6): 3037–3047. Bibcode:1981PhRvB..24.3037P. doi:10.1103/physrevb.24.3037. ISSN 0163-1829.
- Finnis, M. W.; Sinclair, J. E. (1984). "A simple empirical N-body potential for transition metals". Philosophical Magazine A. 50 (1): 45–55. Bibcode:1984PMagA..50...45F. doi:10.1080/01418618408244210. ISSN 0141-8610.
- "Erratum". Philosophical Magazine A. 53 (1): 161. 1986. Bibcode:1986PMagA..53..161.. doi:10.1080/01418618608242815. ISSN 0141-8610.
- Cleri F, Rosato V (July 1993). "Tight-binding potentials for transition metals and alloys". Physical Review B. 48 (1): 22–33. Bibcode:1993PhRvB..48...22C. doi:10.1103/physrevb.48.22. PMID 10006745.
- Kelchner, Cynthia L.; Halstead, David M.; Perkins, Leslie S.; Wallace, Nora M.; DePristo, Andrew E. (1994). "Construction and evaluation of embedding functions". Surface Science. 310 (1–3): 425–435. Bibcode:1994SurSc.310..425K. doi:10.1016/0039-6028(94)91405-2. ISSN 0039-6028.
- Dudarev, S L; Derlet, P M (17 October 2005). "A 'magnetic' interatomic potential for molecular dynamics simulations". Journal of Physics: Condensed Matter. 17 (44): 7097–7118. Bibcode:2005JPCM...17.7097D. doi:10.1088/0953-8984/17/44/003. ISSN 0953-8984.
- Olsson, Pär; Wallenius, Janne; Domain, Christophe; Nordlund, Kai; Malerba, Lorenzo (21 December 2005). "Two-band modeling of α-prime phase formation in Fe-Cr". Physical Review B. 72 (21): 214119. Bibcode:2005PhRvB..72u4119O. doi:10.1103/physrevb.72.214119. ISSN 1098-0121. S2CID 16118006.
- Tersoff J (April 1988). "New empirical approach for the structure and energy of covalent systems". Physical Review B. 37 (12): 6991–7000. Bibcode:1988PhRvB..37.6991T. doi:10.1103/PhysRevB.37.6991. PMID 9943969.
- Brenner DW (November 1990). "Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films". Physical Review B. 42 (15): 9458–9471. Bibcode:1990PhRvB..42.9458B. doi:10.1103/PhysRevB.42.9458. PMID 9995183.
- Brenner DW (August 1989). "Relationship between the embedded-atom method and Tersoff potentials". Physical Review Letters. 63 (9): 1022. Bibcode:1989PhRvL..63.1022B. doi:10.1103/PhysRevLett.63.1022. PMID 10041250.
- Albe, Karsten; Nordlund, Kai; Averback, Robert S. (2002). "Modeling the metal-semiconductor interaction: Analytical bond-order potential for platinum-carbon". Physical Review B. 65 (19): 195124. Bibcode:2002PhRvB..65s5124A. doi:10.1103/PhysRevB.65.195124. ISSN 0163-1829.
- de Brito Mota, F.; Justo, J. F.; Fazzio, A. (1998). "Structural properties of amorphous silicon nitride". Phys. Rev. B. 58 (13): 8323. Bibcode:1998PhRvB..58.8323D. doi:10.1103/PhysRevB.58.8323.
- Juslin, N.; Erhart, P.; Träskelin, P.; Nord, J.; Henriksson, K. O. E.; Nordlund, K.; Salonen, E.; Albe, K. (15 December 2005). "Analytical interatomic potential for modeling nonequilibrium processes in the W–C–H system". Journal of Applied Physics. 98 (12): 123520–123520–12. Bibcode:2005JAP....98l3520J. doi:10.1063/1.2149492. ISSN 0021-8979. S2CID 8090449.
- Erhart, Paul; Juslin, Niklas; Goy, Oliver; Nordlund, Kai; Müller, Ralf; Albe, Karsten (30 June 2006). "Analytic bond-order potential for atomistic simulations of zinc oxide". Journal of Physics: Condensed Matter. 18 (29): 6585–6605. Bibcode:2006JPCM...18.6585E. doi:10.1088/0953-8984/18/29/003. ISSN 0953-8984.
- Baskes MI (December 1987). "Application of the embedded-atom method to covalent materials: A semiempirical potential for silicon". Physical Review Letters. 59 (23): 2666–2669. Bibcode:1987PhRvL..59.2666B. doi:10.1103/PhysRevLett.59.2666. PMID 10035617.
- Baskes MI (August 1992). "Modified embedded-atom potentials for cubic materials and impurities". Physical Review B. 46 (5): 2727–2742. Bibcode:1992PhRvB..46.2727B. doi:10.1103/PhysRevB.46.2727. PMID 10003959.
- Lee, Byeong-Joo; Baskes, M. I. (2000-10-01). "Second nearest-neighbor modified embedded-atom-method potential". Physical Review B. 62 (13): 8564–8567. Bibcode:2000PhRvB..62.8564L. doi:10.1103/PhysRevB.62.8564.
- Heinz H, Lin TJ, Mishra RK, Emami FS (February 2013). "Thermodynamically consistent force fields for the assembly of inorganic, organic, and biological nanostructures: the INTERFACE force field". Langmuir. 29 (6): 1754–65. doi:10.1021/la3038846. PMID 23276161.
- Heinz H, Ramezani-Dakhel H (January 2016). "Simulations of inorganic-bioorganic interfaces to discover new materials: insights, comparisons to experiment, challenges, and opportunities". Chemical Society Reviews. 45 (2): 412–48. doi:10.1039/c5cs00890e. PMID 26750724.
- Mishra, Ratan K.; Mohamed, Aslam Kunhi; Geissbühler, David; Manzano, Hegoi; Jamil, Tariq; Shahsavari, Rouzbeh; Kalinichev, Andrey G.; Galmarini, Sandra; Tao, Lei; Heinz, Hendrik; Pellenq, Roland (December 2017). "A force field database for cementitious materials including validations, applications and opportunities". Cement and Concrete Research. 102: 68–89. doi:10.1016/j.cemconres.2017.09.003.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (July 2004). "Development and testing of a general amber force field". Journal of Computational Chemistry. 25 (9): 1157–74. doi:10.1002/jcc.20035. PMID 15116359.
- Huang J, MacKerell AD (September 2013). "CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data". Journal of Computational Chemistry. 34 (25): 2135–45. doi:10.1002/jcc.23354. PMC 3800559. PMID 23832629.
- Bartók, Albert P.; Kondor, Risi; Csányi, Gábor (2013-05-28). "On representing chemical environments". Physical Review B. 87 (18): 184115. arXiv:1209.3140. Bibcode:2013PhRvB..87r4115B. doi:10.1103/PhysRevB.87.184115. ISSN 1098-0121. S2CID 118375156.
- Deringer, Volker L.; Csányi, Gábor (2017-03-03). "Machine learning based interatomic potential for amorphous carbon". Physical Review B. 95 (9): 094203. arXiv:1611.03277. Bibcode:2017PhRvB..95i4203D. doi:10.1103/PhysRevB.95.094203. ISSN 2469-9950. S2CID 55190594.
- Behler J, Parrinello M (April 2007). "Generalized neural-network representation of high-dimensional potential-energy surfaces". Physical Review Letters. 98 (14): 146401. Bibcode:2007PhRvL..98n6401B. doi:10.1103/PhysRevLett.98.146401. PMID 17501293.
- Bartók AP, Payne MC, Kondor R, Csányi G (April 2010). "Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons". Physical Review Letters. 104 (13): 136403. arXiv:0910.1019. Bibcode:2010PhRvL.104m6403B. doi:10.1103/PhysRevLett.104.136403. PMID 20481899. S2CID 15918457.
- Dragoni, Daniele; Daff, Thomas D.; Csányi, Gábor; Marzari, Nicola (2018-01-30). "Achieving DFT accuracy with a machine-learning interatomic potential: Thermomechanics and defects in bcc ferromagnetic iron". Physical Review Materials. 2 (1): 013808. doi:10.1103/PhysRevMaterials.2.013808. hdl:10281/231112.
- Thompson, A.P.; Swiler, L.P.; Trott, C.R.; Foiles, S.M.; Tucker, G.J. (2015-03-15). "Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials". Journal of Computational Physics. 285: 316–330. arXiv:1409.3880. Bibcode:2015JCoPh.285..316T. doi:10.1016/j.jcp.2014.12.018.
- Byggmästar, J.; Hamedani, A.; Nordlund, K.; Djurabekova, F. (2019-10-17). "Machine-learning interatomic potential for radiation damage and defects in tungsten". Physical Review B. 100 (14): 144105. arXiv:1908.07330. Bibcode:2019PhRvB.100n4105B. doi:10.1103/PhysRevB.100.144105. hdl:10138/306660. S2CID 201106123.
- Pun GP, Batra R, Ramprasad R, Mishin Y (May 2019). "Physically informed artificial neural networks for atomistic modeling of materials". Nature Communications. 10 (1): 2339. Bibcode:2019NatCo..10.2339P. doi:10.1038/s41467-019-10343-5. PMC 6538760. PMID 31138813.
- Heinz, Hendrik; Vaia, R. A.; Farmer, B. L.; Naik, R. R. (2008-10-09). "Accurate Simulation of Surfaces and Interfaces of Face-Centered Cubic Metals Using 12−6 and 9−6 Lennard-Jones Potentials". The Journal of Physical Chemistry C. 112 (44): 17281–17290. doi:10.1021/jp801931d. ISSN 1932-7447.
- Liu, Juan; Tennessen, Emrys; Miao, Jianwei; Huang, Yu; Rondinelli, James M.; Heinz, Hendrik (2018-05-31). "Understanding Chemical Bonding in Alloys and the Representation in Atomistic Simulations". The Journal of Physical Chemistry C. 122 (26): 14996–15009. doi:10.1021/acs.jpcc.8b01891. ISSN 1932-7447.
- Nathanson M, Kanhaiya K, Pryor A, Miao J, Heinz H (December 2018). "Atomic-Scale Structure and Stress Release Mechanism in Core-Shell Nanoparticles". ACS Nano. 12 (12): 12296–12304. doi:10.1021/acsnano.8b06118. PMID 30457827.
- Ruiz, Victor G.; Liu, Wei; Tkatchenko, Alexandre (2016-01-15). "Density-functional theory with screened van der Waals interactions applied to atomic and molecular adsorbates on close-packed and non-close-packed surfaces". Physical Review B. 93 (3): 035118. Bibcode:2016PhRvB..93c5118R. doi:10.1103/physrevb.93.035118. hdl:11858/00-001M-0000-0029-3035-8. ISSN 2469-9950.
- Ruiz VG, Liu W, Zojer E, Scheffler M, Tkatchenko A (April 2012). "Density-functional theory with screened van der Waals interactions for the modeling of hybrid inorganic-organic systems". Physical Review Letters. 108 (14): 146103. Bibcode:2012PhRvL.108n6103R. doi:10.1103/physrevlett.108.146103. PMID 22540809.
- Ercolessi, F; Adams, J. B (10 June 1994). "Interatomic Potentials from First-Principles Calculations: The Force-Matching Method". Europhysics Letters (EPL). 26 (8): 583–588. arXiv:cond-mat/9306054. Bibcode:1994EL.....26..583E. doi:10.1209/0295-5075/26/8/005. ISSN 0295-5075. S2CID 18043298.
- Mishin, Y.; Mehl, M. J.; Papaconstantopoulos, D. A. (12 June 2002). "Embedded-atom potential forB2−NiAl". Physical Review B. 65 (22): 224114. Bibcode:2002PhRvB..65v4114M. doi:10.1103/physrevb.65.224114. ISSN 0163-1829.
- Beardmore, Keith; Smith, Roger (1996). "Empirical potentials for C-Si-H systems with application to C60 interactions with Si crystal surfaces". Philosophical Magazine A. 74 (6): 1439–1466. Bibcode:1996PMagA..74.1439B. doi:10.1080/01418619608240734. ISSN 0141-8610.
- Mishra, Ratan K.; Flatt, Robert J.; Heinz, Hendrik (2013-04-19). "Force Field for Tricalcium Silicate and Insight into Nanoscale Properties: Cleavage, Initial Hydration, and Adsorption of Organic Molecules". The Journal of Physical Chemistry C. 117 (20): 10417–10432. doi:10.1021/jp312815g. ISSN 1932-7447.
- Ramezani-Dakhel, Hadi; Ruan, Lingyan; Huang, Yu; Heinz, Hendrik (2015-01-21). "Molecular Mechanism of Specific Recognition of Cubic Pt Nanocrystals by Peptides and of the Concentration-Dependent Formation from Seed Crystals". Advanced Functional Materials. 25 (9): 1374–1384. doi:10.1002/adfm.201404136. ISSN 1616-301X.
- Chen J, Zhu E, Liu J, Zhang S, Lin Z, Duan X, et al. (December 2018). "Building two-dimensional materials one row at a time: Avoiding the nucleation barrier". Science. 362 (6419): 1135–1139. Bibcode:2018Sci...362.1135C. doi:10.1126/science.aau4146. PMID 30523105. S2CID 54456982.
- Swamy, Varghese; Gale, Julian D. (1 August 2000). "Transferable variable-charge interatomic potential for atomistic simulation of titanium oxides". Physical Review B. 62 (9): 5406–5412. Bibcode:2000PhRvB..62.5406S. doi:10.1103/physrevb.62.5406. ISSN 0163-1829.
- Aguado, Andrés; Bernasconi, Leonardo; Madden, Paul A. (2002). "A transferable interatomic potential for MgO from ab initio molecular dynamics". Chemical Physics Letters. 356 (5–6): 437–444. Bibcode:2002CPL...356..437A. doi:10.1016/s0009-2614(02)00326-3. ISSN 0009-2614.
- Technology, U.S. Department of Commerce, National Institute of Standards and. "Interatomic Potentials Repository Project". www.ctcms.nist.gov.
- "Open Knowledgebase of Interatomic Models (OpenKIM)".
- Acevedo O, Jorgensen WL (January 2010). "Advances in quantum and molecular mechanical (QM/MM) simulations for organic and enzymatic reactions". Accounts of Chemical Research. 43 (1): 142–51. doi:10.1021/ar900171c. PMC 2880334. PMID 19728702.