Density functional theory
Density-functional theory (DFT) is a computational quantum mechanical modelling method used in physics, chemistry and materials science to investigate the electronic structure (or nuclear structure) (principally the ground state) of many-body systems, in particular atoms, molecules, and the condensed phases. Using this theory, the properties of a many-electron system can be determined by using functionals, i.e. functions of another function. In the case of DFT, these are functionals of the spatially dependent electron density. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry.
DFT has been very popular for calculations in solid-state physics since the 1970s. However, DFT was not considered accurate enough for calculations in quantum chemistry until the 1990s, when the approximations used in the theory were greatly refined to better model the exchange and correlation interactions. Computational costs are relatively low when compared to traditional methods, such as exchange only Hartree–Fock theory and its descendants that include electron correlation. Since, DFT has become an important tool for methods of nuclear spectroscopy such as Mössbauer spectroscopy or perturbed angular correlation, in order to understand the reason of specific electric field gradients in crystals.
Despite recent improvements, there are still difficulties in using density functional theory to properly describe: intermolecular interactions (of critical importance to understanding chemical reactions), especially van der Waals forces (dispersion); charge transfer excitations; transition states, global potential energy surfaces, dopant interactions and some strongly correlated systems; and in calculations of the band gap and ferromagnetism in semiconductors.[1] The incomplete treatment of dispersion can adversely affect the accuracy of DFT (at least when used alone and uncorrected) in the treatment of systems which are dominated by dispersion (e.g. interacting noble gas atoms)[2] or where dispersion competes significantly with other effects (e.g. in biomolecules).[3] The development of new DFT methods designed to overcome this problem, by alterations to the functional[4] or by the inclusion of additive terms,[5][6][7][8] is a current research topic. Classical density functional theory uses a similar formalism to calculate properties of non-uniform classical fluids.
Despite the current popularity of these alterations or of the inclusion of additional terms, they are reported[9] to stray away from the search for the exact functional. Further, DFT potentials obtained with adjustable parameters are no longer true DFT potentials,[10] given that they are not functional derivatives of the exchange correlation energy with respect to the charge density. Consequently, it is not clear if the second theorem of DFT holds[10][11] in such conditions.
Overview of method
In the context of computational materials science, ab initio (from first principles) DFT calculations allow the prediction and calculation of material behavior on the basis of quantum mechanical considerations, without requiring higher-order parameters such as fundamental material properties. In contemporary DFT techniques the electronic structure is evaluated using a potential acting on the system's electrons. This DFT potential is constructed as the sum of external potentials Vext, which is determined solely by the structure and the elemental composition of the system, and an effective potential Veff, which represents interelectronic interactions. Thus, a problem for a representative supercell of a material with n electrons can be studied as a set of n one-electron Schrödinger-like equations, which are also known as Kohn–Sham equations.[12]
Origins
Although density functional theory has its roots in the Thomas–Fermi model for the electronic structure of materials, DFT was first put on a firm theoretical footing by Walter Kohn and Pierre Hohenberg in the framework of the two Hohenberg–Kohn theorems (HK).[13] The original HK theorems held only for non-degenerate ground states in the absence of a magnetic field, although they have since been generalized to encompass these.[14][15]
The first HK theorem demonstrates that the ground-state properties of a many-electron system are uniquely determined by an electron density that depends on only three spatial coordinates. It set down the groundwork for reducing the many-body problem of N electrons with 3N spatial coordinates to three spatial coordinates, through the use of functionals of the electron density. This theorem has since been extended to the time-dependent domain to develop time-dependent density functional theory (TDDFT), which can be used to describe excited states.
The second HK theorem defines an energy functional for the system and proves that the ground-state electron density minimizes this energy functional.
In work that later won them the Nobel prize in chemistry, the HK theorem was further developed by Walter Kohn and Lu Jeu Sham to produce Kohn–Sham DFT (KS DFT). Within this framework, the intractable many-body problem of interacting electrons in a static external potential is reduced to a tractable problem of noninteracting electrons moving in an effective potential. The effective potential includes the external potential and the effects of the Coulomb interactions between the electrons, e.g., the exchange and correlation interactions. Modeling the latter two interactions becomes the difficulty within KS DFT. The simplest approximation is the local-density approximation (LDA), which is based upon exact exchange energy for a uniform electron gas, which can be obtained from the Thomas–Fermi model, and from fits to the correlation energy for a uniform electron gas. Non-interacting systems are relatively easy to solve, as the wavefunction can be represented as a Slater determinant of orbitals. Further, the kinetic energy functional of such a system is known exactly. The exchange–correlation part of the total energy functional remains unknown and must be approximated.
Another approach, less popular than KS DFT but arguably more closely related to the spirit of the original HK theorems, is orbital-free density functional theory (OFDFT), in which approximate functionals are also used for the kinetic energy of the noninteracting system.
Derivation and formalism
As usual in many-body electronic structure calculations, the nuclei of the treated molecules or clusters are seen as fixed (the Born–Oppenheimer approximation), generating a static external potential V, in which the electrons are moving. A stationary electronic state is then described by a wavefunction Ψ(r1, …, rN) satisfying the many-electron time-independent Schrödinger equation

where, for the N-electron system, Ĥ is the Hamiltonian, E is the total energy, T̂ is the kinetic energy, V̂ is the potential energy from the external field due to positively charged nuclei, and Û is the electron–electron interaction energy. The operators T̂ and Û are called universal operators, as they are the same for any N-electron system, while V̂ is system-dependent. This complicated many-particle equation is not separable into simpler single-particle equations because of the interaction term Û.
There are many sophisticated methods for solving the many-body Schrödinger equation based on the expansion of the wavefunction in Slater determinants. While the simplest one is the Hartree–Fock method, more sophisticated approaches are usually categorized as post-Hartree–Fock methods. However, the problem with these methods is the huge computational effort, which makes it virtually impossible to apply them efficiently to larger, more complex systems.
Here DFT provides an appealing alternative, being much more versatile, as it provides a way to systematically map the many-body problem, with Û, onto a single-body problem without Û. In DFT the key variable is the electron density n(r), which for a normalized Ψ is given by

This relation can be reversed, i.e., for a given ground-state density n0(r) it is possible, in principle, to calculate the corresponding ground-state wavefunction Ψ0(r1, …, rN). In other words, Ψ is a unique functional of n0,[13]

and consequently the ground-state expectation value of an observable Ô is also a functional of n0:

In particular, the ground-state energy is a functional of n0:

where the contribution of the external potential can be written explicitly in terms of the ground-state density :



More generally, the contribution of the external potential can be written explicitly in terms of the density :



The functionals T[n] and U[n] are called universal functionals, while V[n] is called a non-universal functional, as it depends on the system under study. Having specified a system, i.e., having specified V̂, one then has to minimize the functional

with respect to n(r), assuming one has reliable expressions for T[n] and U[n]. A successful minimization of the energy functional will yield the ground-state density n0 and thus all other ground-state observables.
The variational problems of minimizing the energy functional E[n] can be solved by applying the Lagrangian method of undetermined multipliers.[16] First, one considers an energy functional that does not explicitly have an electron–electron interaction energy term,

where T̂ denotes the kinetic-energy operator, and V̂s is an effective potential in which the particles are moving. Based on , Kohn–Sham equations of this auxiliary noninteracting system can be derived:


which yields the orbitals φi that reproduce the density n(r) of the original many-body system

The effective single-particle potential can be written as

where is the external potential, the second term is the Hartree term describing the electron–electron Coulomb repulsion, and the last term VXC is the exchange–correlation potential. Here, VXC includes all the many-particle interactions. Since the Hartree term and VXC depend on n(r), which depends on the φi, which in turn depend on Vs, the problem of solving the Kohn–Sham equation has to be done in a self-consistent (i.e., iterative) way. Usually one starts with an initial guess for n(r), then calculates the corresponding Vs and solves the Kohn–Sham equations for the φi. From these one calculates a new density and starts again. This procedure is then repeated until convergence is reached. A non-iterative approximate formulation called Harris functional DFT is an alternative approach to this.

- Notes
- The one-to-one correspondence between electron density and single-particle potential is not so smooth. It contains kinds of non-analytic structure. Es[n] contains kinds of singularities, cuts and branches. This may indicate a limitation of our hope for representing exchange–correlation functional in a simple analytic form.
- It is possible to extend the DFT idea to the case of the Green function G instead of the density n. It is called as Luttinger–Ward functional (or kinds of similar functionals), written as E[G]. However, G is determined not as its minimum, but as its extremum. Thus we may have some theoretical and practical difficulties.
- There is no one-to-one correspondence between one-body density matrix n(r, r′) and the one-body potential V(r, r′). (Remember that all the eigenvalues of n(r, r′) are 1.) In other words, it ends up with a theory similar to the Hartree–Fock (or hybrid) theory.
Relativistic density functional theory (ab initio functional forms)
The same theorems can be proven in the case of relativistic electrons, thereby providing generalization of DFT for the relativistic case. Unlike the nonrelativistic theory, in the relativistic case it is possible to derive a few exact and explicit formulas for the relativistic density functional.
Let one consider an electron in a hydrogen-like ion obeying the relativistic Dirac equation. The Hamiltonian H for a relativistic electron moving in the Coulomb potential can be chosen in the following form (atomic units are used):

where V = −eZ/r is the Coulomb potential of a pointlike nucleus, p is a momentum operator of the electron, and e, m and c are the elementary charge, electron mass and the speed of light respectively, and finally α and β are a set of Dirac 2 × 2 matrices:

To find out the eigenfunctions and corresponding energies, one solves the eigenfunction equation

where Ψ = (Ψ(1), Ψ(2), Ψ(3), Ψ(4))T is a four-component wavefunction, and E is the associated eigenenergy. It is demonstrated in Brack (1983)[17] that application of the virial theorem to the eigenfunction equation produces the following formula for the eigenenergy of any bound state:

and analogously, the virial theorem applied to the eigenfunction equation with the square of the Hamiltonian[18] yields

It is easy to see that both of the above formulae represent density functionals. The former formula can be easily generalized for the multi-electron case.[19]
One may observe that both of the functionals written above do not have extremals, of course, if a reasonably wide set of functions is allowed for variation. Nevertheless, it is possible to design a density functional with desired extremal properties out of those ones. Let us make it in the following way:

where ne in Kronecker delta symbol of the second term denotes any extremal for the functional represented by the first term of the functional F. The second term amounts to zero for any function that is not an extremal for the first term of functional F. To proceed further we'd like to find Lagrange equation for this functional. In order to do this, we should allocate a linear part of functional increment when the argument function is altered:

Deploying written above equation, it is easy to find the following formula for functional derivative:

where A = mc2∫ ne dτ, and B = √m2c4 + emc2∫Vne dτ, and V(τ0) is a value of potential at some point, specified by support of variation function δn, which is supposed to be infinitesimal. To advance toward Lagrange equation, we equate functional derivative to zero and after simple algebraic manipulations arrive to the following equation:

Apparently, this equation could have solution only if A = B. This last condition provides us with Lagrange equation for functional F, which could be finally written down in the following form:

Solutions of this equation represent extremals for functional F. It's easy to see that all real densities, that is, densities corresponding to the bound states of the system in question, are solutions of written above equation, which could be called the Kohn–Sham equation in this particular case. Looking back onto the definition of the functional F, we clearly see that the functional produces energy of the system for appropriate density, because the first term amounts to zero for such density and the second one delivers the energy value.
Approximations (exchange–correlation functionals)
The major problem with DFT is that the exact functionals for exchange and correlation are not known, except for the free-electron gas. However, approximations exist which permit the calculation of certain physical quantities quite accurately.[20] One of the simplest approximations is the local-density approximation (LDA), where the functional depends only on the density at the coordinate where the functional is evaluated:

The local spin-density approximation (LSDA) is a straightforward generalization of the LDA to include electron spin:

In LDA, the exchange–correlation energy is typically separated into the exchange part and the correlation part: εXC = εX + εC. The exchange part is called the Dirac (or sometimes Slater) exchange, which takes the form εX ∝ n1/3. There are, however, many mathematical forms for the correlation part. Highly accurate formulae for the correlation energy density εC(n↑, n↓) have been constructed from quantum Monte Carlo simulations of jellium.[21] A simple first-principles correlation functional has been recently proposed as well.[22][23] Although unrelated to the Monte Carlo simulation, the two variants provide comparable accuracy.[24]
The LDA assumes that the density is the same everywhere. Because of this, the LDA has a tendency to underestimate the exchange energy and over-estimate the correlation energy.[25] The errors due to the exchange and correlation parts tend to compensate each other to a certain degree. To correct for this tendency, it is common to expand in terms of the gradient of the density in order to account for the non-homogeneity of the true electron density. This allows corrections based on the changes in density away from the coordinate. These expansions are referred to as generalized gradient approximations (GGA)[26][27][28] and have the following form:

Using the latter (GGA), very good results for molecular geometries and ground-state energies have been achieved.
Potentially more accurate than the GGA functionals are the meta-GGA functionals, a natural development after the GGA (generalized gradient approximation). Meta-GGA DFT functional in its original form includes the second derivative of the electron density (the Laplacian), whereas GGA includes only the density and its first derivative in the exchange–correlation potential.
Functionals of this type are, for example, TPSS and the Minnesota Functionals. These functionals include a further term in the expansion, depending on the density, the gradient of the density and the Laplacian (second derivative) of the density.
Difficulties in expressing the exchange part of the energy can be relieved by including a component of the exact exchange energy calculated from Hartree–Fock theory. Functionals of this type are known as hybrid functionals.
Generalizations to include magnetic fields
The DFT formalism described above breaks down, to various degrees, in the presence of a vector potential, i.e. a magnetic field. In such a situation, the one-to-one mapping between the ground-state electron density and wavefunction is lost. Generalizations to include the effects of magnetic fields have led to two different theories: current density functional theory (CDFT) and magnetic field density functional theory (BDFT). In both these theories, the functional used for the exchange and correlation must be generalized to include more than just the electron density. In current density functional theory, developed by Vignale and Rasolt,[15] the functionals become dependent on both the electron density and the paramagnetic current density. In magnetic field density functional theory, developed by Salsbury, Grayce and Harris,[29] the functionals depend on the electron density and the magnetic field, and the functional form can depend on the form of the magnetic field. In both of these theories it has been difficult to develop functionals beyond their equivalent to LDA, which are also readily implementable computationally.
Applications
In general, density functional theory finds increasingly broad application in chemistry and materials science for the interpretation and prediction of complex system behavior at an atomic scale. Specifically, DFT computational methods are applied for synthesis-related systems and processing parameters. In such systems, experimental studies are often encumbered by inconsistent results and non-equilibrium conditions. Examples of contemporary DFT applications include studying the effects of dopants on phase transformation behavior in oxides, magnetic behavior in dilute magnetic semiconductor materials, and the study of magnetic and electronic behavior in ferroelectrics and dilute magnetic semiconductors.[1][30] It has also been shown that DFT gives good results in the prediction of sensitivity of some nanostructures to environmental pollutants like sulfur dioxide[31] or acrolein,[32] as well as prediction of mechanical properties.[33]
In practice, Kohn–Sham theory can be applied in several distinct ways, depending on what is being investigated. In solid-state calculations, the local density approximations are still commonly used along with plane-wave basis sets, as an electron-gas approach is more appropriate for electrons delocalised through an infinite solid. In molecular calculations, however, more sophisticated functionals are needed, and a huge variety of exchange–correlation functionals have been developed for chemical applications. Some of these are inconsistent with the uniform electron-gas approximation; however, they must reduce to LDA in the electron-gas limit. Among physicists, one of the most widely used functionals is the revised Perdew–Burke–Ernzerhof exchange model (a direct generalized gradient parameterization of the free-electron gas with no free parameters); however, this is not sufficiently calorimetrically accurate for gas-phase molecular calculations. In the chemistry community, one popular functional is known as BLYP (from the name Becke for the exchange part and Lee, Yang and Parr for the correlation part). Even more widely used is B3LYP, which is a hybrid functional in which the exchange energy, in this case from Becke's exchange functional, is combined with the exact energy from Hartree–Fock theory. Along with the component exchange and correlation funсtionals, three parameters define the hybrid functional, specifying how much of the exact exchange is mixed in. The adjustable parameters in hybrid functionals are generally fitted to a "training set" of molecules. Although the results obtained with these functionals are usually sufficiently accurate for most applications, there is no systematic way of improving them (in contrast to some of the traditional wavefunction-based methods like configuration interaction or coupled cluster theory). In the current DFT approach it is not possible to estimate the error of the calculations without comparing them to other methods or experiments.
Thomas–Fermi model
The predecessor to density functional theory was the Thomas–Fermi model, developed independently by both Llewellyn Thomas and Enrico Fermi in 1927. They used a statistical model to approximate the distribution of electrons in an atom. The mathematical basis postulated that electrons are distributed uniformly in phase space with two electrons in every of volume.[34] For each element of coordinate space volume we can fill out a sphere of momentum space up to the Fermi momentum [35]




Equating the number of electrons in coordinate space to that in phase space gives

Solving for pF and substituting into the classical kinetic energy formula then leads directly to a kinetic energy represented as a functional of the electron density:

where

As such, they were able to calculate the energy of an atom using this kinetic-energy functional combined with the classical expressions for the nucleus–electron and electron–electron interactions (which can both also be represented in terms of the electron density).
Although this was an important first step, the Thomas–Fermi equation's accuracy is limited because the resulting kinetic-energy functional is only approximate, and because the method does not attempt to represent the exchange energy of an atom as a conclusion of the Pauli principle. An exchange-energy functional was added by Paul Dirac in 1928.
However, the Thomas–Fermi–Dirac theory remained rather inaccurate for most applications. The largest source of error was in the representation of the kinetic energy, followed by the errors in the exchange energy, and due to the complete neglect of electron correlation.
Edward Teller (1962) showed that Thomas–Fermi theory cannot describe molecular bonding. This can be overcome by improving the kinetic-energy functional.
The kinetic-energy functional can be improved by adding the von Weizsäcker (1935) correction:[36][37]

Hohenberg–Kohn theorems
The Hohenberg–Kohn theorems relate to any system consisting of electrons moving under the influence of an external potential.
Theorem 1. The external potential (and hence the total energy), is a unique functional of the electron density.
- If two systems of electrons, one trapped in a potential and the other in , have the same ground-state density , then is necessarily a constant.




- Corollary: the ground-state density uniquely determines the potential and thus all properties of the system, including the many-body wavefunction. In particular, the HK functional, defined as , is a universal functional of the density (not depending explicitly on the external potential).
- Corollary 2: In light of the fact that the sum of the occupied energies provides the energy content of the Hamiltonian, a unique functional of the ground state charge density, the spectrum of the Hamiltonian is also a unique functional of the ground state charge density.[11]

Theorem 2. The functional that delivers the ground-state energy of the system gives the lowest energy if and only if the input density is the true ground-state density.
- In other words, the energy content of the Hamiltonian reaches its absolute minimum, i.e., the ground state, when the charge density is that of the ground state.
- For any positive integer and potential , a density functional exists such that
- reaches its minimal value at the ground-state density of electrons in the potential . The minimal value of is then the ground-state energy of this system.





Pseudo-potentials
The many-electron Schrödinger equation can be very much simplified if electrons are divided in two groups: valence electrons and inner core electrons. The electrons in the inner shells are strongly bound and do not play a significant role in the chemical binding of atoms; they also partially screen the nucleus, thus forming with the nucleus an almost inert core. Binding properties are almost completely due to the valence electrons, especially in metals and semiconductors. This separation suggests that inner electrons can be ignored in a large number of cases, thereby reducing the atom to an ionic core that interacts with the valence electrons. The use of an effective interaction, a pseudopotential, that approximates the potential felt by the valence electrons, was first proposed by Fermi in 1934 and Hellmann in 1935. In spite of the simplification pseudo-potentials introduce in calculations, they remained forgotten until the late 1950s.
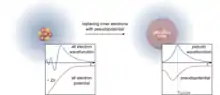
Ab initio pseudo-potentials
A crucial step toward more realistic pseudo-potentials was given by Topp and Hopfield[38] and more recently Cronin, who suggested that the pseudo-potential should be adjusted such that they describe the valence charge density accurately. Based on that idea, modern pseudo-potentials are obtained inverting the free-atom Schrödinger equation for a given reference electronic configuration and forcing the pseudo-wavefunctions to coincide with the true valence wavefunctions beyond a certain distance rl. The pseudo-wavefunctions are also forced to have the same norm (i.e., the so-called norm-conserving condition) as the true valence wavefunctions and can be written as

where Rl(r) is the radial part of the wavefunction with angular momentum l, and PP and AE denote the pseudo-wavefunction and the true (all-electron) wavefunction respectively. The index n in the true wavefunctions denotes the valence level. The distance rl beyond which the true and the pseudo-wavefunctions are equal is also dependent on l.
Electron smearing
The electrons of a system will occupy the lowest Kohn–Sham eigenstates up to a given energy level according to the Aufbau principle. This corresponds to the steplike Fermi–Dirac distribution at absolute zero. If there are several degenerate or close to degenerate eigenstates at the Fermi level, it is possible to get convergence problems, since very small perturbations may change the electron occupation. One way of damping these oscillations is to smear the electrons, i.e. allowing fractional occupancies.[39] One approach of doing this is to assign a finite temperature to the electron Fermi–Dirac distribution. Other ways is to assign a cumulative Gaussian distribution of the electrons or using a Methfessel–Paxton method.[40][41]
Software supporting DFT
DFT is supported by many quantum-chemistry and solid-state physics software packages, often along with other methods.
Classical density functional theory
Classical density functional theory is a classical statistical method to investigate the properties of many-body systems consisting of interacting molecules, macromolecules, nanoparticles or microparticles.[42][43][44][45] The classical non-relativistic method is correct for classical fluids with particle velocities less than the speed of light and thermal de Broglie wavelength smaller than the distance between particles. The theory is based on the calculus of variations of a thermodynamic functional, which is a function of the spatially dependent density function of particles, thus the name. The same name is used for quantum DFT, which is the theory to calculate the electronic structure of electrons based on spatially dependent electron density with quantum and relativistic effects. Classical DFT is a popular and useful method to study fluid phase transitions, ordering in complex liquids, physical characteristics of interfaces and nanomaterials. Since the 1970s it has been applied to the fields of materials science, biophysics, chemical engineering and civil engineering.[46] Computational costs are much lower than for molecular dynamics simulations, which provide similar data and a more detailed description but are limited to small systems and short time scales. Classical DFT is valuable to interpret and test numerical results and to define trends although details of the precise motion of the particles are lost due to averaging over all possible particle trajectories.[47] As in electronic systems, there are fundamental and numerical difficulties in using DFT to quantitatively describe the effect of intermolecular interaction on structure, correlations and thermodynamic properties.
Classical DFT addresses the difficulty of describing thermodynamic equilibrium states of many-particle systems with nonuniform density.[48] Classical DFT has its roots in theories such as the van der Waals theory for the equation of state and the virial expansion method for the pressure. In order to account for correlation in the positions of particles the direct correlation function was introduced as the effective interaction between two particles in the presence of a number of surrounding particles by Leonard Ornstein and Frits Zernike in 1914.[49] The connection to the density pair distribution function was given by the Ornstein–Zernike equation. The importance of correlation for thermodynamic properties was explored through density distribution functions. The functional derivative was introduced to define the distribution functions of classical mechanical systems. Theories were developed for simple and complex liquids using the ideal gas as a basis for the free energy and adding molecular forces as a second-order perturbation. A term in the gradient of the density was added to account for non-uniformity in density in the presence of external fields or surfaces. These theories can be considered precursors of DFT.
To develop a formalism for the statistical thermodynamics of non-uniform fluids functional differentiation was used extensively by Percus and Lebowitz (1961), which led to the Percus–Yevick equation linking the density distribution function and the direct correlation.[50] Other closure relations were also proposed;the Classical-map hypernetted-chain method, the BBGKY hierarchy. In the late 1970s classical DFT was applied to the liquid–vapor interface and the calculation of surface tension. Other applications followed: the freezing of simple fluids, formation of the glass phase, the crystal–melt interface and dislocation in crystals, properties of polymer systems, and liquid crystal ordering. Classical DFT was applied to colloid dispersions, which were discovered to be good models for atomic systems.[51] By assuming local chemical equilibrium and using the local chemical potential of the fluid from DFT as the driving force in fluid transport equations, equilibrium DFT is extended to describe non-equilibrium phenomena and fluid dynamics on small scales.
Classical DFT allows the calculation of the equilibrium particle density and prediction of thermodynamic properties and behavior of a many-body system on the basis of model interactions between particles. The spatially dependent density determines the local structure and composition of the material. It is determined as a function that optimizes the thermodynamic potential of the grand canonical ensemble. The grand potential is evaluated as the sum of the ideal-gas term with the contribution from external fields and an excess thermodynamic free energy arising from interparticle interactions. In the simplest approach the excess free-energy term is expanded on a system of uniform density using a functional Taylor expansion. The excess free energy is then a sum of the contributions from s-body interactions with density-dependent effective potentials representing the interactions between s particles. In most calculations the terms in the interactions of three or more particles are neglected (second-order DFT). When the structure of the system to be studied is not well approximated by a low-order perturbation expansion with a uniform phase as the zero-order term, non-perturbative free-energy functionals have also been developed. The minimization of the grand potential functional in arbitrary local density functions for fixed chemical potential, volume and temperature provides self-consistent thermodynamic equilibrium conditions, in particular, for the local chemical potential. The functional is not in general a convex functional of the density; solutions may not be local minima. Limiting to low-order corrections in the local density is a well-known problem, although the results agree (reasonably) well on comparison to experiment.
A variational principle is used to determine the equilibrium density. It can be shown that for constant temperature and volume the correct equilibrium density minimizes the grand potential functional of the grand canonical ensemble over density functions . In the language of functional differentiation (Mermin theorem):


The Helmholtz free energy functional is defined as . The functional derivative in the density function determines the local chemical potential: . In classical statistical mechanics the partition function is a sum over probability for a given microstate of N classical particles as measured by the Boltzmann factor in the Hamiltonian of the system. The Hamiltonian splits into kinetic and potential energy, which includes interactions between particles, as well as external potentials. The partition function of the grand canonical ensemble defines the grand potential. A correlation function is introduced to describe the effective interaction between particles.



The s-body density distribution function is defined as the statistical ensemble average of particle positions. It measures the probability to find s particles at points in space :



From the definition of the grand potential, the functional derivative with respect to the local chemical potential is the density; higher-order density correlations for two, three, four or more particles are found from higher-order derivatives:

The radial distribution function with s = 2 measures the change in the density at a given point for a change of the local chemical interaction at a distant point.
In a fluid the free energy is a sum of the ideal free energy and the excess free-energy contribution from interactions between particles. In the grand ensemble the functional derivatives in the density yield the direct correlation functions :



The one-body direct correlation function plays the role of an effective mean field. The functional derivative in density of the one-body direct correlation results in the direct correlation function between two particles . The direct correlation function is the correlation contribution to the change of local chemical potential at a point for a density change at and is related to the work of creating density changes at different positions. In dilute gases the direct correlation function is simply the pair-wise interaction between particles (Debye–Huckel equation). The Ornstein–Zernike equation between the pair and the direct correlation functions is derived from the equation




Various assumptions and approximations adapted to the system under study lead to expressions for the free energy. Correlation functions are used to calculate the free-energy functional as an expansion on a known reference system. If the non-uniform fluid can be described by a density distribution that is not far from uniform density a functional Taylor expansion of the free energy in density increments leads to an expression for the thermodynamic potential using known correlation functions of the uniform system. In the square gradient approximation a strong non-uniform density contributes a term in the gradient of the density. In a perturbation theory approach the direct correlation function is given by the sum of the direct correlation in a known system such as hard spheres and a term in a weak interaction such as the long range London dispersion force. In a local density approximation the local excess free energy is calculated from the effective interactions with particles distributed at uniform density of the fluid in a cell surrounding a particle. Other improvements have been suggested such as the weighted density approximation for a direct correlation function of a uniform system which distributes the neighboring particles with an effective weighted density calculated from a self-consistent condition on the direct correlation function.
The variational Mermin principle leads to an equation for the equilibrium density and system properties are calculated from the solution for the density. The equation is a non-linear integro-differential equation and finding a solution is not trivial, requiring numerical methods, except for the simplest models. Classical DFT is supported by standard software packages, and specific software is currently under development. Assumptions can be made to propose trial functions as solutions, and the free energy is expressed in the trial functions and optimized with respect to parameters of the trial functions. Examples are a localized Gaussian function centered on crystal lattice points for the density in a solid, the hyperbolic function for interfacial density profiles.

Classical DFT has found many applications, for example:
- developing new functional materials in materials science, in particular nanotechnology;
- studying the properties of fluids at surfaces and the phenomena of wetting and adsorption;[52]
- understanding life processes in biotechnology;
- improving filtration methods for gases and fluids in chemical engineering;
- fighting pollution of water and air in environmental science;
- generating new procedures in microfluidics and nanofluidics.
The extension of classical DFT towards nonequilibrium systems is known as dynamical density functional theory (DDFT).[53] DDFT allows to describe the time evolution of the one-body density of a colloidal system, which is governed by the equation


with the mobility and the free energy . DDFT can be derived from the microscopic equations of motion for a colloidal system (Langevin equations or Smoluchowski equation) based on the adiabatic approximation, which corresponds to the assumption that the two-body distribution in a nonequilibrium system is identical to that in an equilibrium system with the same one-body density. For a system of noninteracting particles, DDFT reduces to the standard diffusion equation.

See also
- Basis set (chemistry)
- Dynamical mean field theory
- Gas in a box
- Harris functional
- Helium atom
- Kohn–Sham equations
- Local density approximation
- Molecule
- Molecular design software
- Molecular modelling
- Quantum chemistry
- Thomas–Fermi model
- Time-dependent density functional theory
- Car–Parrinello molecular dynamics
Lists
- List of quantum chemistry and solid state physics software
- List of software for molecular mechanics modeling
References
- Assadi, M. H. N.; et al. (2013). "Theoretical study on copper's energetics and magnetism in TiO2 polymorphs". Journal of Applied Physics. 113 (23): 233913–233913–5. arXiv:1304.1854. Bibcode:2013JAP...113w3913A. doi:10.1063/1.4811539. S2CID 94599250.
- Van Mourik, Tanja; Gdanitz, Robert J. (2002). "A critical note on density functional theory studies on rare-gas dimers". Journal of Chemical Physics. 116 (22): 9620–9623. Bibcode:2002JChPh.116.9620V. doi:10.1063/1.1476010.
- Vondrášek, Jiří; Bendová, Lada; Klusák, Vojtěch; Hobza, Pavel (2005). "Unexpectedly strong energy stabilization inside the hydrophobic core of small protein rubredoxin mediated by aromatic residues: correlated ab initio quantum chemical calculations". Journal of the American Chemical Society. 127 (8): 2615–2619. doi:10.1021/ja044607h. PMID 15725017.
- Grimme, Stefan (2006). "Semiempirical hybrid density functional with perturbative second-order correlation". Journal of Chemical Physics. 124 (3): 034108. Bibcode:2006JChPh.124c4108G. doi:10.1063/1.2148954. PMID 16438568. S2CID 28234414.
- Zimmerli, Urs; Parrinello, Michele; Koumoutsakos, Petros (2004). "Dispersion corrections to density functionals for water aromatic interactions". Journal of Chemical Physics. 120 (6): 2693–2699. Bibcode:2004JChPh.120.2693Z. doi:10.1063/1.1637034. PMID 15268413. S2CID 20826940.
- Grimme, Stefan (2004). "Accurate description of van der Waals complexes by density functional theory including empirical corrections". Journal of Computational Chemistry. 25 (12): 1463–1473. doi:10.1002/jcc.20078. PMID 15224390. S2CID 6968902.
- Von Lilienfeld, O. Anatole; Tavernelli, Ivano; Rothlisberger, Ursula; Sebastiani, Daniel (2004). "Optimization of effective atom centered potentials for London dispersion forces in density functional theory" (PDF). Physical Review Letters. 93 (15): 153004. Bibcode:2004PhRvL..93o3004V. doi:10.1103/PhysRevLett.93.153004. PMID 15524874.
- Tkatchenko, Alexandre; Scheffler, Matthias (2009). "Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data". Physical Review Letters. 102 (7): 073005. Bibcode:2009PhRvL.102g3005T. doi:10.1103/PhysRevLett.102.073005. PMID 19257665.
- Medvedev, Michael G.; Bushmarinov, Ivan S.; Sun, Jianwei; Perdew, John P.; Lyssenko, Konstantin A. (2017-01-05). "Density functional theory is straying from the path toward the exact functional". Science. 355 (6320): 49–52. Bibcode:2017Sci...355...49M. doi:10.1126/science.aah5975. ISSN 0036-8075. PMID 28059761. S2CID 206652408.
- Jiang, Hong (2013-04-07). "Band gaps from the Tran-Blaha modified Becke-Johnson approach: A systematic investigation". The Journal of Chemical Physics. 138 (13): 134115. doi:10.1063/1.4798706. ISSN 0021-9606. PMID 23574216.
- Bagayoko, Diola (December 2014). "Understanding density functional theory (DFT) and completing it in practice". AIP Advances. 4 (12): 127104. doi:10.1063/1.4903408. ISSN 2158-3226.
- Hanaor, D. A. H.; Assadi, M. H. N.; Li, S.; Yu, A.; Sorrell, C. C. (2012). "Ab initio study of phase stability in doped TiO2". Computational Mechanics. 50 (2): 185–194. arXiv:1210.7555. Bibcode:2012CompM..50..185H. doi:10.1007/s00466-012-0728-4. S2CID 95958719.
- Hohenberg, Pierre; Walter, Kohn (1964). "Inhomogeneous electron gas". Physical Review. 136 (3B): B864–B871. Bibcode:1964PhRv..136..864H. doi:10.1103/PhysRev.136.B864.
- Levy, Mel (1979). "Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem". Proceedings of the National Academy of Sciences. 76 (12): 6062–6065. Bibcode:1979PNAS...76.6062L. doi:10.1073/pnas.76.12.6062. PMC 411802. PMID 16592733.
- Vignale, G.; Rasolt, Mark (1987). "Density-functional theory in strong magnetic fields". Physical Review Letters. 59 (20): 2360–2363. Bibcode:1987PhRvL..59.2360V. doi:10.1103/PhysRevLett.59.2360. PMID 10035523.
- Kohn, W.; Sham, L. J. (1965). "Self-consistent equations including exchange and correlation effects". Physical Review. 140 (4A): A1133–A1138. Bibcode:1965PhRv..140.1133K. doi:10.1103/PhysRev.140.A1133.
- Brack, M. (1983). "Virial theorems for relativistic spin-1/2 and spin-0 particles" (PDF). Physical Review D. 27 (8): 1950. Bibcode:1983PhRvD..27.1950B. doi:10.1103/physrevd.27.1950.
- Koshelev, K. (2015). "About density functional theory interpretation". arXiv:0812.2919 [quant-ph].
- Koshelev, K. (2007). "Alpha variation problem and q-factor definition". arXiv:0707.1146 [physics.atom-ph].
- Burke, Kieron; Wagner, Lucas O. (2013). "DFT in a nutshell". International Journal of Quantum Chemistry. 113 (2): 96. doi:10.1002/qua.24259.
- Perdew, John P.; Ruzsinszky, Adrienn; Tao, Jianmin; Staroverov, Viktor N.; Scuseria, Gustavo; Csonka, Gábor I. (2005). "Prescriptions for the design and selection of density functional approximations: More constraint satisfaction with fewer fits". Journal of Chemical Physics. 123 (6): 062201. Bibcode:2005JChPh.123f2201P. doi:10.1063/1.1904565. PMID 16122287. S2CID 13097889.
- Chachiyo, Teepanis (2016). "Communication: Simple and accurate uniform electron gas correlation energy for the full range of densities". Journal of Chemical Physics. 145 (2): 021101. Bibcode:2016JChPh.145b1101C. doi:10.1063/1.4958669. PMID 27421388.
- Fitzgerald, Richard J. (2016). "A simpler ingredient for a complex calculation". Physics Today. 69 (9): 20. Bibcode:2016PhT....69i..20F. doi:10.1063/PT.3.3288.
- Jitropas, Ukrit; Hsu, Chung-Hao (2017). "Study of the first-principles correlation functional in the calculation of silicon phonon dispersion curves". Japanese Journal of Applied Physics. 56 (7): 070313. Bibcode:2017JaJAP..56g0313J. doi:10.7567/JJAP.56.070313.
- Becke, Axel D. (2014-05-14). "Perspective: Fifty years of density-functional theory in chemical physics". The Journal of Chemical Physics. 140 (18): A301. Bibcode:2014JChPh.140rA301B. doi:10.1063/1.4869598. ISSN 0021-9606. PMID 24832308. S2CID 33556753.
- Perdew, John P.; Chevary, J. A.; Vosko, S. H.; Jackson, Koblar A.; Pederson, Mark R.; Singh, D. J.; Fiolhais, Carlos (1992). "Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation". Physical Review B. 46 (11): 6671–6687. Bibcode:1992PhRvB..46.6671P. doi:10.1103/physrevb.46.6671. hdl:10316/2535. PMID 10002368.
- Becke, Axel D. (1988). "Density-functional exchange-energy approximation with correct asymptotic behavior". Physical Review A. 38 (6): 3098–3100. Bibcode:1988PhRvA..38.3098B. doi:10.1103/physreva.38.3098. PMID 9900728.
- Langreth, David C.; Mehl, M. J. (1983). "Beyond the local-density approximation in calculations of ground-state electronic properties". Physical Review B. 28 (4): 1809. Bibcode:1983PhRvB..28.1809L. doi:10.1103/physrevb.28.1809.
- Grayce, Christopher; Harris, Robert (1994). "Magnetic-field density-functional theory". Physical Review A. 50 (4): 3089–3095. Bibcode:1994PhRvA..50.3089G. doi:10.1103/PhysRevA.50.3089. PMID 9911249.
- Segall, M. D.; Lindan, P. J. (2002). "First-principles simulation: ideas, illustrations and the CASTEP code". Journal of Physics: Condensed Matter. 14 (11): 2717. Bibcode:2002JPCM...14.2717S. CiteSeerX 10.1.1.467.6857. doi:10.1088/0953-8984/14/11/301.
- Soleymanabadi, Hamed; Rastegar, Somayeh F. (2014-01-01). "Theoretical investigation on the selective detection of SO2 molecule by AlN nanosheets". Journal of Molecular Modeling. 20 (9): 2439. doi:10.1007/s00894-014-2439-6. PMID 25201451. S2CID 26745531.
- Soleymanabadi, Hamed; Rastegar, Somayeh F. (2013-01-01). "DFT studies of acrolein molecule adsorption on pristine and Al-doped graphenes". Journal of Molecular Modeling. 19 (9): 3733–3740. doi:10.1007/s00894-013-1898-5. PMID 23793719. S2CID 41375235.
- Music, D.; Geyer, R. W.; Schneider, J. M. (2016). "Recent progress and new directions in density functional theory based design of hard coatings". Surface & Coatings Technology. 286: 178–190. doi:10.1016/j.surfcoat.2015.12.021.
- Parr & Yang (1989), p. 47.
- March, N. H. (1992). Electron Density Theory of Atoms and Molecules. Academic Press. p. 24. ISBN 978-0-12-470525-8.
- von Weizsäcker, C. F. (1935). "Zur Theorie der Kernmassen" [On the theory of nuclear masses]. Zeitschrift für Physik (in German). 96 (7–8): 431–458. Bibcode:1935ZPhy...96..431W. doi:10.1007/BF01337700. S2CID 118231854.
- Parr & Yang (1989), p. 127.
- Topp, William C.; Hopfield, John J. (1973-02-15). "Chemically Motivated Pseudopotential for Sodium". Physical Review B. 7 (4): 1295–1303. Bibcode:1973PhRvB...7.1295T. doi:10.1103/PhysRevB.7.1295.
- Michelini, M. C.; Pis Diez, R.; Jubert, A. H. (25 June 1998). "A Density Functional Study of Small Nickel Clusters". International Journal of Quantum Chemistry. 70 (4–5): 694. doi:10.1002/(SICI)1097-461X(1998)70:4/5<693::AID-QUA15>3.0.CO;2-3.
- "Finite temperature approaches – smearing methods". VASP the GUIDE. Retrieved 21 October 2016.
- Tong, Lianheng. "Methfessel–Paxton Approximation to Step Function". Metal CONQUEST. Retrieved 21 October 2016.
- Evans, Robert (1979). "The nature of the liquid-vapor interface and other topics in the statistical mechanics of non-uniform classical fluids". Advances in Physics. 281 (2): 143–200. Bibcode:1979AdPhy..28..143E. doi:10.1080/00018737900101365.
- Evans, Robert; Oettel, Martin; Roth, Roland; Kahl, Gerhard (2016). "New developments in classical density functional theory". Journal of Physics: Condensed Matter. 28 (24): 240401. doi:10.1088/0953-8984/28/24/240401. ISSN 0953-8984. PMID 27115564. Retrieved 2020-09-10.
- Singh, Yaswant (1991). "Density Functional Theory of Freezing and Properties of the ordered Phase". Physics Reports. 207 (6): 351–444. Bibcode:1991PhR...207..351S. doi:10.1016/0370-1573(91)90097-6.
- ten Bosch, Alexandra (2019). Analytical Molecular Dynamics:from Atoms to Oceans. ISBN 978-1091719392.
- Wu, Jianzhong (2006). "Density Functional Theory for chemical engineering:from capillarity to soft materials". AIChE Journal. 52 (3): 1169–1193. doi:10.1002/aic.10713.
- Gelb, Lev D.; Gubbins, K. E.; Radhakrishnan, R.; Sliwinska-Bartkowiak, M. (1999). "Phase separation in confined systemss". Reports on Progress in Physics. 62 (12): 1573–1659. Bibcode:1999RPPh...62.1573G. doi:10.1088/0034-4885/62/12/201. S2CID 9282112.
- Frisch, Harry; Lebowitz, Joel (1964). The equilibrium theory of classical fluids. New York: W. A. Benjamin.
- Ornstein, L. S.; Zernike, F. (1914). "Accidental deviations of density and opalescence at the critical point of a single substance" (PDF). Royal Netherlands Academy of Arts and Sciences (KNAW). Proceedings. 17: 793–806. Bibcode:1914KNAB...17..793.
- Lebowitz, J. L.; Percus, J. K. (1963). "Statistical Thermodynamics of Non-uniform Fluids". Journal of Mathematical Physics. 4 (1): 116–123. Bibcode:1963JMP.....4..116L. doi:10.1063/1.1703877.
- Löwen, Hartmut (1994). "Melting, freezing and colloidal suspensions". Physics Reports. 237 (5): 241–324. Bibcode:1994PhR...237..249L. doi:10.1016/0370-1573(94)90017-5.
- Hydrophobicity of ceria, Applied Surface Science, 2019, 478, pp.68-74. in HAL archives ouvertes
- te Vrugt, Michael; Löwen, Hartmut; Wittkowski, Raphael (2020). "Classical dynamical density functional theory: from fundamentals to applications". Advances in Physics. 69 (2): 121–247. arXiv:2009.07977. doi:10.1080/00018732.2020.1854965.
Sources
- Parr, R. G.; Yang, W. (1989). Density-Functional Theory of Atoms and Molecules. New York: Oxford University Press. ISBN 978-0-19-504279-5.
- Thomas, L. H. (1927). "The calculation of atomic fields". Proc. Camb. Phil. Soc. 23 (5): 542–548. Bibcode:1927PCPS...23..542T. doi:10.1017/S0305004100011683. S2CID 122732216.
- Hohenberg, P.; Kohn, W. (1964). "Inhomogeneous Electron Gas". Physical Review. 136 (3B): B864. Bibcode:1964PhRv..136..864H. doi:10.1103/PhysRev.136.B864.
- Kohn, W.; Sham, L. J. (1965). "Self-Consistent Equations Including Exchange and Correlation Effects". Physical Review. 140 (4A): A1133. Bibcode:1965PhRv..140.1133K. doi:10.1103/PhysRev.140.A1133.
- Becke, Axel D. (1993). "Density-functional thermochemistry. III. The role of exact exchange". The Journal of Chemical Physics. 98 (7): 5648. Bibcode:1993JChPh..98.5648B. doi:10.1063/1.464913. S2CID 52389061.
- Lee, Chengteh; Yang, Weitao; Parr, Robert G. (1988). "Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density". Physical Review B. 37 (2): 785–789. Bibcode:1988PhRvB..37..785L. doi:10.1103/PhysRevB.37.785. PMID 9944570. S2CID 45348446.
- Burke, Kieron; Werschnik, Jan; Gross, E. K. U. (2005). "Time-dependent density functional theory: Past, present, and future". The Journal of Chemical Physics. 123 (6): 062206. arXiv:cond-mat/0410362. Bibcode:2005JChPh.123f2206B. doi:10.1063/1.1904586. PMID 16122292. S2CID 2659101.
- Lejaeghere, K.; Bihlmayer, G.; Bjorkman, T.; Blaha, P.; Blugel, S.; Blum, V.; Caliste, D.; Castelli, I. E.; Clark, S. J.; Dal Corso, A.; de Gironcoli, S.; Deutsch, T.; Dewhurst, J. K.; Di Marco, I.; Draxl, C.; Du ak, M.; Eriksson, O.; Flores-Livas, J. A.; Garrity, K. F.; Genovese, L.; Giannozzi, P.; Giantomassi, M.; Goedecker, S.; Gonze, X.; Granas, O.; Gross, E. K. U.; Gulans, A.; Gygi, F.; Hamann, D. R.; Hasnip, P. J.; Holzwarth, N. A. W.; Iu an, D.; Jochym, D. B.; Jollet, F.; Jones, D.; Kresse, G.; Koepernik, K.; Kucukbenli, E.; Kvashnin, Y. O.; Locht, I. L. M.; Lubeck, S.; Marsman, M.; Marzari, N.; Nitzsche, U.; Nordstrom, L.; Ozaki, T.; Paulatto, L.; Pickard, C. J.; Poelmans, W.; Probert, M. I. J.; Refson, K.; Richter, M.; Rignanese, G.-M.; Saha, S.; Scheffler, M.; Schlipf, M.; Schwarz, K.; Sharma, S.; Tavazza, F.; Thunstrom, P.; Tkatchenko, A.; Torrent, M.; Vanderbilt, D.; van Setten, M. J.; Van Speybroeck, V.; Wills, J. M.; Yates, J. R.; Zhang, G.-X.; Cottenier, S. (2016). "Reproducibility in density functional theory calculations of solids". Science. 351 (6280): aad3000. Bibcode:2016Sci...351.....L. doi:10.1126/science.aad3000. ISSN 0036-8075. PMID 27013736.
External links
- Walter Kohn, Nobel Laureate – Video interview with Walter on his work developing density functional theory by the Vega Science Trust
- Capelle, Klaus (2002). "A bird's-eye view of density-functional theory". arXiv:cond-mat/0211443.
- Walter Kohn, Nobel Lecture
- Density functional theory on arxiv.org
- FreeScience Library -> Density Functional Theory
- Argaman, Nathan; Makov, Guy (2000). "Density Functional Theory -- an introduction". American Journal of Physics. 68 (2000): 69–79. arXiv:physics/9806013. Bibcode:2000AmJPh..68...69A. doi:10.1119/1.19375. S2CID 119102923.
- Electron Density Functional Theory – Lecture Notes
- Density Functional Theory through Legendre Transformationpdf
- Burke, Kieron. "The ABC of DFT" (PDF).
- Modeling Materials Continuum, Atomistic and Multiscale Techniques, Book
- NIST Jarvis-DFT
Atomic models | |
|---|---|
| Single atoms |
|
| Atoms in solids | |
| Atoms in fluids | |
| Scientists | |
| |
