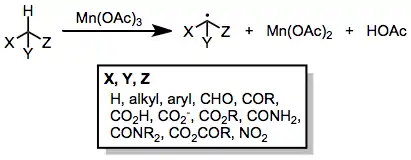
Manganese-mediated coupling reactions
Manganese-mediated coupling reactions are radical coupling reactions between enolizable carbonyl compounds and unsaturated compounds initiated by a manganese(III) salt, typically manganese(III) acetate. Copper(II) acetate is sometimes used as a co-oxidant to assist in the oxidation of intermediate radicals to carbocations.[1]
Introduction
Manganese(III) acetate is a one-electron oxidizing agent that is particularly effective for the oxidation of enolizable carbonyl compounds to α-oxoalkyl or α,α'-dioxoalkyl radicals.[2] Radicals generated in this manner may then undergo inter- or intramolecular addition to carbon-carbon multiple bonds. Pathways available to the adduct radical include further oxidation to a carbocation (and subsequent β-elimination or trapping with a nucleophile) and hydrogen abstraction to generate a saturated carbonyl compound containing a new carbon-carbon bond. Copper(II) acetate is sometimes needed to facilitate the oxidation of adduct radicals to carbocations.[3] Yields of these reactions are generally moderate, particularly in the intermolecular case, but tandem intramolecular radical cyclizations initiated by Mn(III) oxidation may generate complex carbocyclic frameworks. Because of the limited functional group compatibility of Mn(OAc)3, radical couplings employing this reagent have mainly been applied to the synthesis of hydrocarbon natural products, such as pheromones.[4]
(1)
Mechanism and Stereochemistry
Prevailing Mechanism
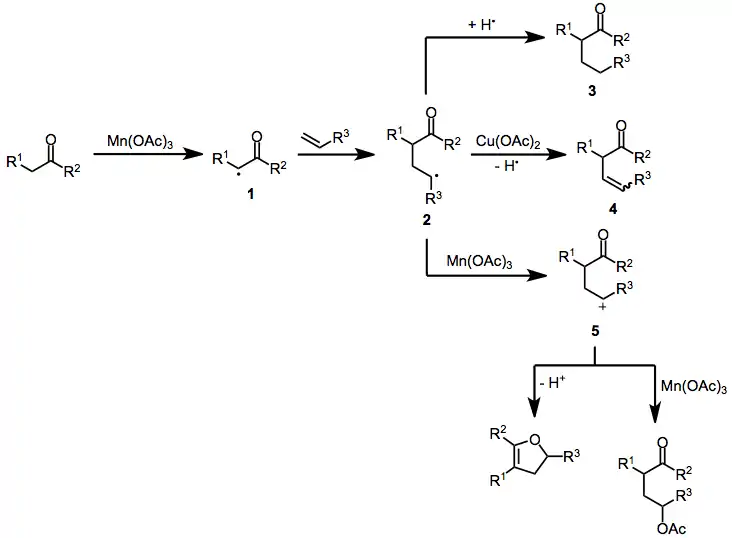
The mechanism of manganese(III)-mediated radical reactions begins with the single-electron oxidation of a carbonyl compound to an α-oxoalkyl radical. Addition to an olefin then occurs, generating adduct radical 2. The fate of 2 is primarily determined by reaction conditions—in the presence of copper(II) acetate, this intermediate undergoes further oxidation to a carbocation and may eliminate to form β,γ-unsaturated ketone 4. Manganese acetate itself can effect the second oxidation of resonance-stabilized adduct radicals to carbocations 5;[5] unstabilized radicals undergo further transformations before reacting with Mn(OAc)3. Atom transfer from another molecule of substrate may generate saturated compound 3. Adduct radicals or carbocations may undergo ligand-transfer reactions, yielding γ-functionalized carbonyl compounds. When lithium chloride is used as an additive, chlorination takes place.[6] Alternatively, carbocations may be trapped intramolecularly by the carbonyl oxygen to form dihydrofurans after β-elimination.[7]
(2)
Scope and Limitations
The outcomes of manganese-mediated coupling reactions depend on both the structure of the substrate(s) and the reaction conditions. This section describes the scope and limitations of inter- and intramolecular manganese-mediated radical coupling reactions and is organized according to the carbonyl compound employed as the substrate.
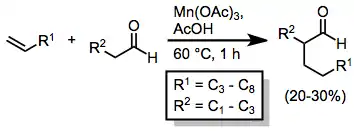
Intermolecular reactions between ketones/aldehydes and alkenes tend to result in low yields. In the absence of copper(II) acetate, hydrogen atom abstraction occurs, yielding saturated ketones or aldehydes.[8]
(3)
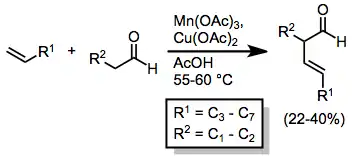
When Cu(OAc)2 is present, further oxidation to carbocations followed by elimination takes place, leading to the formation of β,γ-unsaturated carbonyl compounds in moderate yields.[9]
(4)
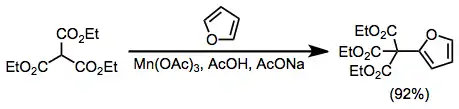
Aromatic compounds are also useful radical acceptors in manganese(III)-mediated coupling reactions. Furan reacts selectively at the α position to afford substituted products in high yield.[10]
(5)
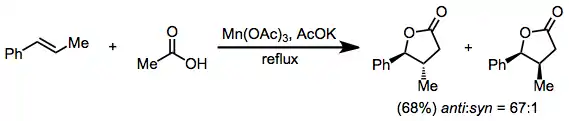
Lactonization of alkenes in the presence of acetic acid and acetate salts is a synthetically useful method for the synthesis of γ-lactones. Selectivity is high for the radical addition that leads to the more stable adduct radical, and trans lactones are selectively formed from either cis or trans acyclic alkenes.[11]
(6)
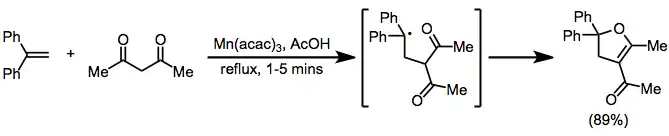
β-Dicarbonyl compounds are useful substrates for the formation of dihydrofurans. Copper(II) acetate is not necessary in this case because of the high resonance stabilization of the intermediate diphenylmethyl radical.[12]
(7)
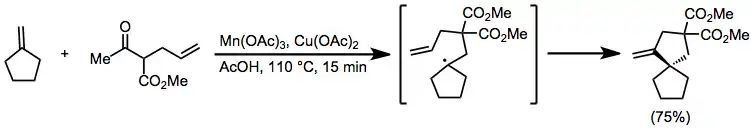
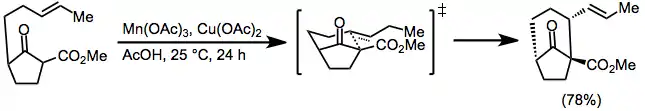
When alkenes or carbonyl compounds containing pendant unsaturated moiety are treated with manganese(III) acetate, tandem intramolecular cyclization reactions may occur. Generally, exo cyclization of terminal double bonds is favored, as shown in equation (10).[13] A variety of substitution patterns may be employed for this transformation, and yields are generally higher than intermolecular coupling reactions.[14]
(8)
The stereochemical course of tandem reactions can be understood in some cases by invoking a chairlike transition state with as many substituents as possible in pseudoequatorial positions;[15] however, a number of examples exhibiting unpredictable stereochemistry are known.[16]
(9)
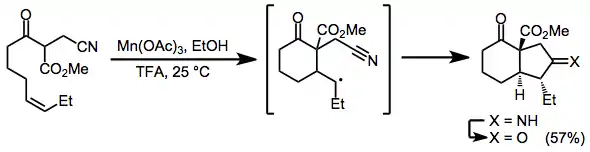
Nitriles are useful as radical acceptors in tandem cyclizations. Hydrolysis of the resulting imine leads to polycyclic ketones in moderate yields with good stereoselectivity.[17]
(10)
Synthetic Applications
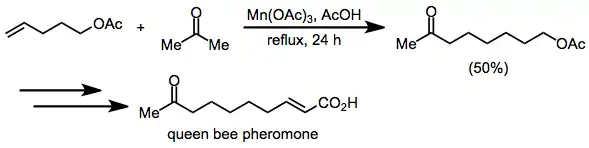
Synthetic applications of manganese-mediated coupling have focused primarily on the synthesis of hydrocarbon natural products, such as pheromones. A synthesis of queen bee pheromone uses the intermolecular coupling of acetone and an ω-alkenyl acetate en route to the target.[18]
(11)
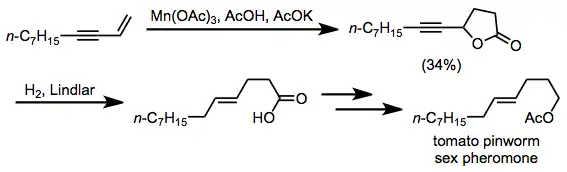
Lactonization is a key step in the synthesis of tomato pinworm sex pheromone. Subsequent Lindlar hydrogenation, reduction and acetylation provided the target compound.[19]
(12)
Experimental Conditions and Procedure
Typical Conditions
Manganese(III) acetate may be used either as its dihydrate or in the anhydrous form. Alternatively, it can be electrochemically generated from manganese(II) acetate.[20] [21] Stoichiometry is important to consider for these reactions, as two equivalents of oxidant are required if carbocation intermediates are desired (because Mn(OAc)3 is a one-electron oxidant). A second equivalent of manganese(III) acetate is also required if copper(II) acetate is used as a co-catalyst, because Mn(OAc)3 re-oxidizes Cu(I) to Cu(II). Glacial acetic acid is the most common solvent used, and although it results in heterogeneous reaction mixtures at room temperature, brief heating can be applied before the substrate is added to dissolve the Mn(OAc)3. Because a variety of paths of different molecularity are open to the intermediates of the reaction, concentration is an important variable to consider. Elevated temperatures are typically required to effect the oxidation of less acidic carbonyl compounds.
Example Procedure[22]
(13)
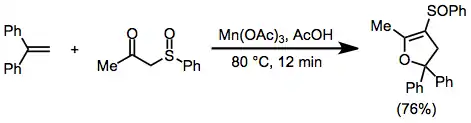
To a heated solution of phenylsulfenylacetone (1 mmol) and 1,1-diphenylethene (1 mmol) in AcOH (25 mL), Mn(OAc)3·2 H2O (4 mmol) was added. The mixture was stirred at 80° for 12 minutes. The reaction was quenched by adding H2O (60 mL), and the mixture was then extracted with benzene. After removing the benzene, the product was separated as a pale yellow oil, either on TLC (Wacogel B10) eluting with CHCl3 or on a silica-gel column eluting with benzene. IR (CHCl3) 1642, 1018 cm−1; 1H NMR (60 MHz, CDCl3) δ2.38 (t, J = 1.6 Hz, 3 H), 2.86 (dq, J = 1.6, 14.4 Hz, 1 H), 3.72 (dq, J = 1.6, 14.4 Hz, 1 H), 7.07–7.73 (m, 15 H); high-resolution MS m/e Calcd for C23H20O2S 360.1184. Found 360.1273.
References
- Melikyan, G. G. Org. React. 1997, 49, 427. doi:10.1002/0471264180.or049.03
- Snider, B. B. Chemtracts-Org. Chem. 1991, 4, 403.
- Vinogradov, M. G.; Il'ina, G.; Nikishin, G. I. J. Org. Chem. USSR 1974, 10, 1167.
- Melikyan, G. G.; Mkrtchyan, V. M.; Atanesyan, K. A.; Asaryan, G. Kh.; Badanyan, Sh. O. Chem. Nat. Compd. 1990, 78.
- Melikyan, G. G.; Vostrowsky, O.; Bauer, W.; Bestmann, H. J.; Khan, M.; Nicholas, K. M. J. Org. Chem. 1994, 59, 222.
- Vinogradov, M. G.; Dolinko, V. I.; Nikishin, G. I. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1984, 334.
- Qian, C.; Nishino, H.; Kurosawa, K. J. Heterocycl. Chem. 1993, 30, 209.
- Nikishin, G. I.; Vinogradov, M. G.; Il'ina, G. J. Org. Chem. USSR 1972, 8, 1422.
- Nikishin, G. I.; Vinogradov, M. G.; Il'ina, G. P. Synthesis 1972, 376.
- Cho, I.; Muchowski, J. Synthesis 1991, 567.
- Fristad, W. E.; Peterson, J. R. J. Org. Chem. 1985, 50, 10.
- Yang, F. Z.; Trost, M. K.; Fristad, W. E. Tetrahedron Lett. 1987, 28, 1493.
- McQuillin, F. J.; Wood, M. J. Chem. Soc., Perkin Trans. 1 1976, 1762.
- Snider, B. B.; Dombroski, M. A. J. Org. Chem. 1987, 52, 5487.
- Kates, S. A.; Dombroski, M. A.; Snider, B. B. J. Org. Chem. 1990, 55, 2427.
- Bertrand, M. P.; Surzur, J.-M.; Oumar-Mahamat, H.; Moustrou, C. J. Org. Chem. 1991, 56, 3089.
- Snider, B. B.; Buckman, B. O. J. Org. Chem. 1992, 57, 322.
- Subramanian, C. S.; Thomas, P. J.; Mamdapur, V. R.; Chadha, M. S. Indian J. Chem. 1978, 16B, 318.
- Melikyan, G. G.; Aslanyan, G. Kh.; Atanesyan, K. A.; Mkrtchyan, D. A.; Badanyan, Sh. O. Chem. Nat. Compd. 1990, 83.
- Bellami, A. J. Acta Chem. Scand. 1979, B33, 208.
- Yılmaz, M.; Yılmaz, E. V. B.; Pekel, A. T. Helv. Chim. Acta 2011, 94, 2027-2038. doi:10.1002/hlca.201100105
- Qian, C.; Nishino, H.; Kurosawa, K. J. Heterocycl. Chem. 1993, 30, 209.