Muir–Torre syndrome
Muir–Torre syndrome is a rare hereditary, autosomal dominant cancer syndrome[1]:663 that is thought to be a subtype of HNPCC. Individuals are prone to develop cancers of the colon, genitourinary tract, and skin lesions, such as keratoacanthomas and sebaceous tumors. The genes affected are MLH1, MSH2, and more recently, MSH6, and are involved in DNA mismatch repair.
| Muir–Torre syndrome | |
|---|---|
| Other names | MTS |
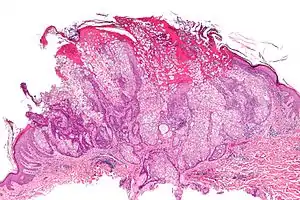 | |
| Micrograph of a sebaceous adenoma, as may be seen in Muir–Torre syndrome. H&E stain. | |
| Specialty | Oncology, dermatology, medical genetics |
Symptoms
Muir–Torre syndrome is characterized by both:[2]
- At least a single sebaceous gland tumor (either an adenoma, an epithelioma, or a carcinoma)
- A minimum of one internal malignancy
The Amsterdam criteria are frequently used to diagnose Lynch syndrome and Muir–Torre syndrome. They include the following:
- 3 or more relatives with an HNPCC-associated cancer (i.e., colorectal, cancer of the endometrium, small bowel, ureter, or renal pelvis)
- 2 or more successive generations affected by cancer
- 1 or more persons with cancer is a first-degree relative of the other 2, at least 1 case of colorectal cancer younger than age 50 years, a diagnosis of familial adenomatous polyposis has been excluded, tumors are verified by histologic examination
Muir–Torre syndrome is a genetic condition. Mutations in MLH1 and MSH2 are linked with the disease. These genes code for DNA mismatch repair genes, and mutations increase the risk of developing cancerous qualities.
Many patients who have sebaceous neoplasms with mutations in MSH2 and MLH1 do not in fact have Muir–Torre syndrome. The Mayo Muir–Torre risk score was devised to improve the positive predictive value of immunohistochemistry and reduce the false positive rate.[3][4] The Mayo Muir–Torre Risk score assigns points based several characteristics. A score of 2 or greater has a high positive predictive value of Muir–Torre syndrome. A score of 1 or lower is less likely to be Muir–Torre syndrome.[3]
Age of onset of first sebaceous neoplasm: <60 years = 1 point, otherwise 0 points Total number of sebaceous neoplasms: 1 = 0 points, >2 = 2 points. Personal history of Lynch related cancers: No = 0 points, Yes = 1 point Family history of Lynch-related cancer: No = 0 points, Yes = 1 point
The most common internal malignancies associated with Muir–Torre syndrome are: Colorectal (56%), Urogenital (22%), Small Intestine (4%), and Breast (4%). A variety of other internal malignancies have been reported.[5]
Cause
Genetic overlap with Turcot syndrome
A couple studies have been conducted on patients with both Muir–Torre syndrome and Turcot syndrome. It is thought that the two may have some genetic overlap. Both have been associated defects in MLH1 and MSH2 genes.[6]
In one study, a patient with defective MSH2 and MSH6 mismatch repair genes exhibited both syndromes. This is the first case where a patient with genotypic changes consistent with HNPCC has been properly diagnosed with an overlap of both syndromes. Along with neoplasms of the sebaceous gland, this patient developed cerebral neoplasms, characteristic of Turcot syndrome.[7]
Treatment
Immunohistochemistry is now being used more often to diagnose patients likely to have Muir–Torre syndrome. Sebaceous neoplasms are only infrequently encountered, and immunohistochemistry is reliable and readily available, so researchers have recommended its use. Routine immunohistochemical detection of DNA mismatch repair proteins help identify hereditary DNA mismatch repair deficiency.[8]
Treatment of Muir–Torre syndrome normally consists of oral isotretinoin. The drug has been found to prevent tumor development.[9][10]
Patients with Muir–Torre syndrome should follow the same stringent screening for colorectal carcinoma and other malignancies as patients with Lynch syndrome. This includes frequent and early colonoscopies, mammograms, dermatologic evaluation, and imaging of the abdomen and pelvis.[11]
Epidemiology
Muir–Torre was observed to occur in 14 of 50 families (28%) and in 14 of 152 individuals (9.2%) with Lynch syndrome, also known as HNPCC.[12]
The 2 major MMR proteins involved are hMLH1 and hMSH2. Approximately 70% of tumors associated with the MTS have microsatellite instability. While germline disruption of hMLH1 and hMSH2 is evenly distributed in HNPCC, disruption of hMSH2 is seen in greater than 90% of MTS patients.[13]
Gastrointestinal and genitourinary cancers are the most common internal malignancies. Colorectal cancer is the most common visceral neoplasm in Muir–Torre syndrome patients.[14]
Eponym
It is named for EG Muir and D Torre. A British physician, Muir noted a patient with many keratoacanthomas who went on to develop several internal malignancies at a young age. Torre presented his findings at a meeting of the New York Dermatologic Society.[15][16]
It was not until the 1980s when Creighton professor Henry Lynch noted a clustering of Muir–Torre syndrome patients in families with Lynch syndrome.[17]
See also
- List of cutaneous conditions
- List of cutaneous conditions associated with increased risk of nonmelanoma skin cancer
References
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- Cohen PR, Kohn R, Kurzrock R (May 1991). "Association of sebaceous gland tumors and internal malignancy: The Muir–Torre syndrome". The American Journal of Medicine. 90 (5): 606–13. doi:10.1016/S0002-9343(05)80013-0. PMID 2029018.
- Roberts ME, Riegert-Johnson DL, Thomas BC; et al. (2014). "A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir–Torre variant of Lynch syndrome". Genetics in Medicine. 16 (9): 711–16. doi:10.1038/gim.2014.19. PMID 24603434.CS1 maint: multiple names: authors list (link)
- Roberts ME, Riegert-Johnson DL, Thomas BC; et al. (2013). "Screening for Muir–Torre syndrome using mismatch repair protein immunohisochemistry of sebaceous neoplasms". J Genet Counsel. 22 (3): 393–405. doi:10.1007/s10897-012-9552-4. PMID 23212176.CS1 maint: multiple names: authors list (link)
- Akhtar S, Oza KK, Khan SA; et al. (1999). "Muir–Torre syndrome: a case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature". J Am Acad Dermatol. 41 (5): 681–6. doi:10.1016/s0190-9622(99)70001-0. PMID 10534628.CS1 maint: multiple names: authors list (link)
- Kleinerman R, Marino J, Loucas E (May 2012). "Muir–Torre Syndrome / Turcot Syndrome overlap? A patient with sebaceous carcinoma, colon cancer, and a malignant astrocytoma". Dermatology Online Journal. 18 (5): 3. PMID 22630573.
- Grandhi R, Deibert CP, Pirris SM, Lembersky B, Mintz AH (April 2013). "Simultaneous Muir–Torre and Turcot's syndrome: A case report and review of the literature". Surg Neurol Int. 4 (52): 52. doi:10.4103/2152-7806.110512. PMC 3640225. PMID 23646262.
- Orta L, Klimstra DS, Qin J, et al. (2009). "Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient's age or other clinical characteristics". Am J Surg Pathol. 33 (6): 934–44. doi:10.1097/PAS.0b013e318199edca. PMID 19342947.
- Graefe T, Wollina U, Schulz H, Burgdorf W (March 2000). "Muir–Torre Syndrome – Treatment with Isotretinoin and Interferon Alpha-2a Can Prevent Tumour Development". Dermatology. 200 (4): 331–3. doi:10.1159/000018399. PMID 10894967.
- Spielvogel RL, DeVillez RL, Roberts LC (March 1985). "Oral isotretinoin therapy for familial Muir–Torre syndrome". J Am Acad Dermatol. 12 (3): 475–80. doi:10.1016/S0190-9622(85)70066-7. PMID 3857234.
- Ponti G, Pnz de Leon M (2005). "Muir–Torre syndrome". Lancet Oncol. 6 (12): 980–87. doi:10.1016/S1470-2045(05)70465-4. PMID 16321766.
- South CD, Hampel H, Comeas I, Westman JA, Frankel WL, Chapelle A (February 2008). "The Frequency of Muir–Torre Syndrome Among Lynch Syndrome Families". Oxford Journal. 100 (4): 277–81. doi:10.1093/jnci/djm291. PMID 18270343.
- Ponti G, Losi L, Pedroni M (October 2006). "Value of MLH1 and MSH2 mutations in the appearance of Muir–Torre syndrome phenotype in HNPCC patients presenting sebaceous gland tumors or keratoacanthomas. |". J Invest Dermatol. 126 (10): 2302–7. doi:10.1038/sj.jid.5700475. PMID 16826164.
- Okan G, Vural P, Ince U, Yazar A, Uras C, Saruc M (August 2012). "Muir–Torre syndrome: a case report and review of the literature". Turk J Gastroenterol. 23 (4): 394–8. doi:10.4318/tjg.2012.0411. PMID 22965514.
- Muir EG, Bell AJ, Barlow KA (March 1967). "Multiple primary carcinomata of the colon, duodenum, and larynx associated with kerato-acanthomata of the face". Br J Surg. 54 (3): 191–5. doi:10.1002/bjs.1800540309. PMID 6020987.
- Torre D (November 1968). "Multiple sebaceous tumors". Arch Dermatol. 98 (5): 549–51. doi:10.1001/archderm.98.5.549. PMID 5684233.
- Lynch HT, Fussaro RM, Roberts L; et al. (1985). "Muir–Torre syndrome in several members of a family with a variant of the Cancer Family Syndrome". Br J Dermatol. 113 (3): 295–301. doi:10.1111/j.1365-2133.1985.tb02081.x. PMID 4063166.CS1 maint: multiple names: authors list (link)
External links
| Classification | |
|---|---|
| External resources |