Orotic aciduria
Orotic aciduria (AKA hereditary orotic aciduria) is a disease caused by an enzyme deficiency resulting in a decreased ability to synthesize pyrimidines. It is the only known enzyme deficiency of the de novo pyrimidine synthesis pathway.[2]
| Orotic aciduria | |
|---|---|
| Other names | Orotidylic pyrophosphorylase and orotidylic decarboxylase deficiency; Uridine monophosphate synthase (UMPS) deficiency[1] |
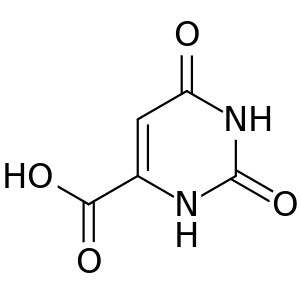 | |
| Structure of orotic acid | |
| Specialty | Hematology |
| Symptoms | Megaloblastic anemia; developmental delays |
| Causes | Autosomal recessive mutation of the UMPS gene |
| Differential diagnosis | Mitochondrial disorders; Lysinuric protein intolerance; liver disease[1] |
| Treatment | Uridine triacetate |
Orotic aciduria is characterized by excessive excretion of orotic acid in urine because of the inability to convert orotic acid to UMP.[3][1] It causes megaloblastic anemia and may be associated with mental and physical developmental delays.
Signs and symptoms
Patients typically present with excessive orotic acid in the urine, failure to thrive, developmental delay, and megaloblastic anemia which cannot be cured by administration of vitamin B12 or folic acid.[3][2]
Cause and genetics
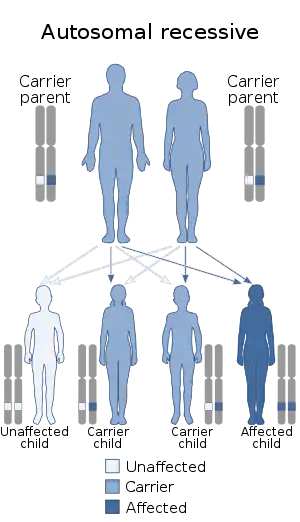
This autosomal recessive disorder is caused by a deficiency in the enzyme UMPS,[4] a bifunctional protein that includes the enzyme activities of OPRT and ODC.[5] In one study of three patients, UMPS activity ranged from 2-7% of normal levels.[2]
Two types of orotic aciduria have been reported. Type I has a severe deficiency of both activities of UMP synthase. In Type II orotic aciduria, the ODC activity is deficient while OPRT activity is elevated. As of 1988, only one case of type II orotic aciduria had ever been reported.[2]
Orotic aciduria is associated with megaloblastic anemia due to decreased pyrimidine synthesis, which leads to decreased nucleotide-lipid cofactors needed for erythrocyte membrane synthesis in the bone marrow.[6]
Diagnosis
Elevated urinary orotic acid levels can also arise secondary to blockage of the urea cycle, particularly in ornithine transcarbamylase deficiency (OTC deficiency). This can be distinguished from hereditary orotic aciduria by assessing blood ammonia levels and blood urea nitrogen (BUN). In OTC deficiency, hyperammonemia and decreased BUN are seen because the urea cycle is not functioning properly, but megaloblastic anemia will not occur because pyrimidine synthesis is not affected.[7] In orotic aciduria, the urea cycle is not affected.
Orotic aciduria can be diagnosed through genetic sequencing of the UMPS gene.[1]
Treatment
Treatment is administration of uridine monophosphate (UMP) or uridine triacetate (which is converted to UMP). These medications will bypass the missing enzyme and provide the body with a source of pyrimidines.[3][1]
References
- "Orotic aciduria type 1". National Center for Advancing Translational Sciences. 13 Sep 2017. Retrieved 8 May 2018.
- Winkler, JK; Suttle, DP (July 1988). "Analysis of UMP synthase gene and mRNA structure in hereditary orotic aciduria fibroblasts". American Journal of Human Genetics. 43 (1): 86–94. PMC 1715274. PMID 2837086.
- Tao, Le (2017-01-02). First aid for the USMLE step 1 2017 : a student-to-student guide. Bhushan, Vikas,, Sochat, Matthew,, Kallianos, Kimberly,, Chavda, Yash,, Zureick, Andrew H. (Andrew Harrison), 1991-, Kalani, Mehboob. New York. ISBN 9781259837630. OCLC 948547794.
- Suchi M, Mizuno H, Kawai Y, Tsuboi T, Sumi S, Okajima K, Hodgson ME, Ogawa H, Wada Y (Mar 1997). "Molecular cloning of the human UMP synthase gene and characterization of point mutations in two hereditary orotic aciduria families". American Journal of Human Genetics. 60 (3): 525–539. ISSN 0002-9297. PMC 1712531. PMID 9042911.
- Donald., Voet (2013). Fundamentals of biochemistry : life at the molecular level. Voet, Judith G., Pratt, Charlotte W. (Fourth ed.). Hoboken, NJ: Wiley. ISBN 9780470547847. OCLC 738349533.
- Balasubramaniam, S; Duley, JA; Christodoulou, J (Sep 2014). "Inborn errors of pyrimidine metabolism: clinical update and therapy". Journal of Inherited Metabolic Disease. 37 (5): 687–98. doi:10.1007/s10545-014-9742-3. PMID 25030255.
- Wraith, J. E. (2001). "Ornithine carbamoyltransferase deficiency". Archives of Disease in Childhood. 84 (1): 84–88. doi:10.1136/adc.84.1.84. PMC 1718609. PMID 11124797.
External links
| Classification |
|---|
