Pathogenomics
Pathogenomics is a field which uses high-throughput screening technology and bioinformatics to study encoded microbe resistance, as well as virulence factors (VFs), which enable a microorganism to infect a host and possibly cause disease.[1][2][3][4] This includes studying genomes of pathogens which cannot be cultured outside of a host.[5] In the past, researchers and medical professionals found it difficult to study and understand pathogenic traits of infectious organisms.[6] With newer technology, pathogen genomes can be identified and sequenced in a much shorter time and at a lower cost,[7][8] thus improving the ability to diagnose, treat, and even predict and prevent pathogenic infections and disease.[9] It has also allowed researchers to better understand genome evolution events - gene loss, gain, duplication, rearrangement - and how those events impact pathogen resistance and ability to cause disease.[8] This influx of information has created a need for making the vast amounts of data accessible to researchers in the form of databases,[10] and it has raised ethical questions about the wisdom of reconstructing previously extinct and deadly pathogens in order to better understand virulence.[11]
History
During the earlier times when genomics was being studied, scientists found it challenging to sequence genetic information.[12] The field began to explode in 1977 when Fred Sanger, PhD, along with his colleagues, sequenced the DNA-based genome of a bacteriophage, using a method now known as the Sanger Method.[13][14][15] The Sanger Method for sequencing DNA exponentially advanced molecular biology and directly led to the ability to sequence genomes of other organisms, including the complete human genome.[13][14]
The Haemophilus influenza genome was one of the first organism genomes sequenced in 1995 by J. Craig Venter and Hamilton Smith using whole genome shotgun sequencing.[16][14] Since then, newer and more efficient high-throughput sequencing, such as Next Generation Genomic Sequencing (NGS) and Single-Cell Genomic Sequencing, have been developed.[14] While the Sanger method is able to sequence one DNA fragment at a time, NGS technology can sequence thousands of sequences at a time.[17] With the ability to rapidly sequence DNA, new insights developed, such as the discovery that since prokaryotic genomes are more diverse than originally thought, it is necessary to sequence multiple strains in a species rather than only a few.[18] E.coli was an example of why this is important, with genes encoding virulence factors in two strains of the species differing by at least thirty percent.[18] Such knowledge, along with more thorough study of genome gain, loss, and change, is giving researchers valuable insight into how pathogens interact in host environments and how they are able to infect hosts and cause disease.[18][12]
With this high influx of new information, there has arisen a higher demand for bioinformatics so scientists can properly analyze the new data. In response, software and other tools have been developed for this purpose.[19] Also, as of 2008, the amount of stored sequences was doubling every 18 months, making urgent the need for better ways to organize data and aid research.[20] In response, thousands of publicly accessible databases and other resources have been created, including the Virulence Factor Database (VFDB) of pathogenic bacteria, which was established in 2004 and was created to aid in pathogenomics research.[21][3][20]
Microbe analysis
Pathogens may be prokaryotic (archaea or bacteria), single-celled eukarya or viruses. Prokaryotic genomes have typically been easier to sequence due to smaller genome size compared to Eukarya. Due to this, there is a bias in reporting pathogenic bacterial behavior. Regardless of this bias in reporting, many of the dynamic genomic events are similar across all the types of pathogen organisms. Genomic evolution occurs via gene gain, gene loss, and genome rearrangement, and these "events" are observed in multiple pathogen genomes, with some bacterial pathogens experiencing all three.[12] Pathogenomics does not focus exclusively on understanding pathogen-host interactions, however. Insight of individual or cooperative pathogen behavior provides knowledge into the development or inheritance of pathogen virulence factors.[12] Through a deeper understanding of the small sub-units that cause infection, it may be possible to develop novel therapeutics that are efficient and cost-effective.[22]
Cause and analysis of genomic diversity
Dynamic genomes with high plasticity are necessary to allow pathogens, especially bacteria, to survive in changing environments.[18] With the assistance of high throughput sequencing methods and in silico technologies, it is possible to detect, compare and catalogue many of these dynamic genomic events. Genomic diversity is important when detecting and treating a pathogen since these events can change the function and structure of the pathogen.[23][24] There is a need to analyze more than a single genome sequence of a pathogen species to understand pathogen mechanisms. Comparative genomics is a methodology which allows scientists to compare the genomes of different species and strains.[25] There are several examples of successful comparative genomics studies, among them the analysis of Listeria[26] and Escherichia coli.[27] Some studies have attempted to address the difference between pathogenic and non-pathogenic microbes. This inquiry proves to be difficult, however, since a single bacterial species can have many strains, and the genomic content of each of these strains varies.[27]
Evolutionary dynamics
Varying microbe strains and genomic content are caused by different forces, including three specific evolutionary events which have an impact on pathogen resistance and ability to cause disease, a: gene gain, gene loss, and genome rearrangement.[12]
Gene loss and genome decay
Gene loss occurs when genes are deleted. The reason why this occurs is still not fully understood,[28] though it most likely involves adaptation to a new environment or ecological niche.[29][30] Some researchers believe gene loss may actually increase fitness and survival among pathogens.[28] In a new environment, some genes may become unnecessary for survival, and so mutations are eventually "allowed" on those genes until they become inactive "pseudogenes."[29] These pseudogenes are observed in organisms such as Shigella flexneri, Salmonella enterica,[31] and Yersinia pestis.[29] Over time, the pseudogenes are deleted, and the organisms become fully dependent on their host as either endosymbionts or obligate intracellular pathogens, as is seen in Buchnera, Myobacterium leprae, and Chlamydia trachomatis.[29] These deleted genes are also called Anti-virulence genes (AVG) since it is thought they may have prevented the organism from becoming pathogenic.[29] In order to be more virulent, infect a host and remain alive, the pathogen had to get rid of those AVGs.[29] The reverse process can happen as well, as was seen during analysis of Listeria strains, which showed that a reduced genome size led to a non-pathogenic Listeria strain from a pathogenic strain.[26] Systems have been developed to detect these pseudogenes/AVGs in a genome sequence.[8]
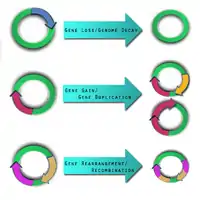
Gene gain and duplication
One of the key forces driving gene gain is thought to be horizontal (lateral) gene transfer (LGT).[32] It is of particular interest in microbial studies because these mobile genetic elements may introduce virulence factors into a new genome.[33] A comparative study conducted by Gill et al. in 2005 postulated that LGT may have been the cause for pathogen variations between Staphylococcus epidermidis and Staphylococcus aureus.[34] There still, however, remains skepticism about the frequency of LGT, its identification, and its impact.[35] New and improved methodologies have been engaged, especially in the study of phylogenetics, to validate the presence and effect of LGT.[36] Gene gain and gene duplication events are balanced by gene loss, such that despite their dynamic nature, the genome of a bacterial species remains approximately the same size.[37]
Genome rearrangement
Mobile genetic insertion sequences can play a role in genome rearrangement activities.[38] Pathogens that do not live in an isolated environment have been found to contain a large number of insertion sequence elements and various repetitive segments of DNA.[18] The combination of these two genetic elements is thought help mediate homologous recombination. There are pathogens, such as Burkholderia mallei,[39] and Burkholderia pseudomallei[40] which have been shown to exhibit genome-wide rearrangements due to insertion sequences and repetitive DNA segments.[18] At this time, no studies demonstrate genome-wide rearrangement events directly giving rise to pathogenic behavior in a microbe. This does not mean it is not possible. Genome-wide rearrangements do, however, contribute to the plasticity of bacterial genome, which may prime the conditions for other factors to introduce, or lose, virulence factors.[18]
Single-nucleotide polymorphisms
Single Nucleotide Polymorphisms, or SNPs, allow for a wide array of genetic variation among humans as well as pathogens. They allow researchers to estimate a variety of factors: the effects of environmental toxins, how different treatment methods affect the body, and what causes someone's predisposition to illnesses.[41] SNPs play a key role in understanding how and why mutations occur. SNPs also allows for scientists to map genomes and analyze genetic information.[41]
Pan and core genomes
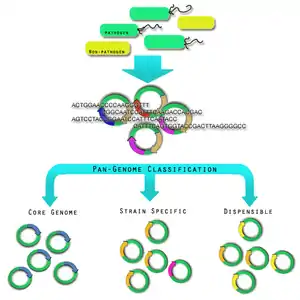
Pan-genome overview The most recent definition of a bacterial species comes from the pre-genomic era. In 1987, it was proposed that bacterial strains showing >70% DNA·DNA re-association and sharing characteristic phenotypic traits should be considered to be strains of the same species.[42] The diversity within pathogen genomes makes it difficult to identify the total number of genes that are associated within all strains of a pathogen species.[42] It has been thought that the total number of genes associated with a single pathogen species may be unlimited,[42] although some groups are attempting to derive a more empirical value.[43] For this reason, it was necessary to introduce the concept of pan-genomes and core genomes.[44] Pan-genome and core genome literature also tends to have a bias towards reporting on prokaryotic pathogenic organisms. Caution may need to be exercised when extending the definition of a pan-genome or a core-genome to the other pathogenic organisms because there is no formal evidence of the properties of these pan-genomes.
A core genome is the set of genes found across all strains of a pathogen species.[42] A pan-genome is the entire gene pool for that pathogen species, and includes genes that are not shared by all strains.[42] Pan-genomes may be open or closed depending on whether comparative analysis of multiple strains reveals no new genes (closed) or many new genes (open) compared to the core genome for that pathogen species.[12] In the open pan-genome, genes may be further characterized as dispensable or strain specific. Dispensable genes are those found in more than one strain, but not in all strains, of a pathogen species.[44] Strain specific genes are those found only in one strain of a pathogen species.[44] The differences in pan-genomes are reflections of the life style of the organism. For example, Streptococcus agalactiae, which exists in diverse biological niches, has a broader pan-genome when compared with the more environmentally isolated Bacillus anthracis.[18] Comparative genomics approaches are also being used to understand more about the pan-genome.[45] Recent discoveries show that the number of new species continue to grow with an estimated 1031 bacteriophages on the planet with those bacteriophages infecting 1024 others per second, the continuous flow of genetic material being exchanged is difficult to imagine.[42]
Virulence factors
Multiple genetic elements of human-affecting pathogens contribute to the transfer of virulence factors: plasmids, pathogenicity island, prophages, bacteriophages, transposons, and integrative and conjugative elements.[12][46] Pathogenicity islands and their detection are the focus of several bioinformatics efforts involved in pathogenomics.[47][48] It is a common belief that "environmental bacterial strains" lack the capacity to harm or do damage to humans. However, recent studies show that bacteria from aquatic environments have acquired pathogenic strains through evolution. This allows for the bacteria to have a wider range in genetic traits and can cause a potential threat to humans from which there is more resistance towards antibiotics.[46]
Microbe-microbe interactions
Microbe-host interactions tend to overshadow the consideration of microbe-microbe interactions. Microbe-microbe interactions though can lead to chronic states of infirmity that are difficult to understand and treat.[9]
Biofilms
Biofilms are an example of microbe-microbe interactions and are thought to be associated with up to 80% of human infections.[49] Recently it has been shown that there are specific genes and cell surface proteins involved in the formation of biofilm.[50] These genes and also surface proteins may be characterized through in silico methods to form an expression profile of biofilm-interacting bacteria.[9] This expression profile may be used in subsequent analysis of other microbes to predict biofilm microbe behaviour, or to understand how to dismantle biofilm formation.[9]
Host microbe analysis
Pathogens have the ability to adapt and manipulate host cells, taking full advantage of a host cell's cellular processes and mechanisms.[9]
A microbe may be influenced by hosts to either adapt to its new environment or learn to evade it. An insight into these behaviours will provide beneficial insight for potential therapeutics. The most detailed outline of host-microbe interaction initiatives is outlined by the Pathogenomics European Research Agenda.[9] Its report emphasizes the following features:
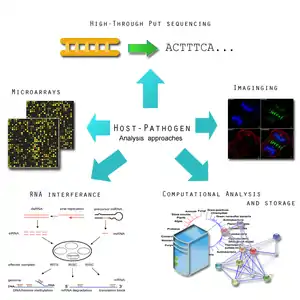
- Microarray analysis of host and microbe gene expression during infection. This is important for identifying the expression of virulence factors that allow a pathogen to survive a host's defense mechanism.[9] Pathogens tend to undergo an assortment of changed in order to subvert and hosts immune system, in some case favoring a hyper variable genome state.[51] The genomic expression studies will be complemented with protein-protein interaction networks studies.[9]
- Using RNA interference (RNAi) to identify host cell functions in response to infections. Infection depends on the balance between the characteristics of the host cell and the pathogen cell. In some cases, there can be an overactive host response to infection, such as in meningitis, which can overwhelm the host's body.[9] Using RNA, it will be possible to more clearly identify how a host cell defends itself during times of acute or chronic infection.[52] This has also been applied successfully is Drosophila.[52]
- Not all microbe interactions in host environment are malicious. Commensal flora, which exists in various environments in animals and humans may actually help combating microbial infections.[9] The human flora, such as the gut for example, is home to a myriad of microbes.[53]
The diverse community within the gut has been heralded to be vital for human health. There are a number of projects under way to better understand the ecosystems of the gut.[54] The sequence of commensal Escherichia coli strain SE11, for example, has already been determined from the faecal matter of a healthy human and promises to be the first of many studies.[55] Through genomic analysis and also subsequent protein analysis, a better understanding of the beneficial properties of commensal flora will be investigated in hopes of understanding how to build a better therapeutic.[56]
Eco-evo perspective
The "eco-evo" perspective on pathogen-host interactions emphasizes the influences ecology and the environment on pathogen evolution.[12] The dynamic genomic factors such as gene loss, gene gain and genome rearrangement, are all strongly influenced by changes in the ecological niche where a particular microbial strain resides. Microbes may switch from being pathogenic and non-pathogenic due to changing environments.[26] This was demonstrated during studies of the plague, Yersinia pestis, which apparently evolved from a mild gastrointestinal pathogen to a very highly pathogenic microbe through dynamic genomic events.[57] In order for colonization to occur, there must be changes in biochemical makeup to aid survival in a variety of environments. This is most likely due to a mechanism allowing the cell to sense changes within the environment, thus influencing change in gene expression.[58] Understanding how these strain changes occur from being low or non-pathogenic to being highly pathogenic and vice versa may aid in developing novel therapeutics for microbial infections.[12]
Applications
Human health has greatly improved and the mortality rate has declined substantially since the second world war because of improved hygiene due to changing public health regulations, as well as more readily available vaccines and antibiotics.[59] Pathogenomics will allow scientists to expand what they know about pathogenic and non-pathogenic microbes, thus allowing for new and improved vaccines.[59] Pathogenomics also has wider implication, including preventing bioterrorism.[59]
Reverse vaccinology
Reverse vaccinology is relatively new. While research is still being conducted, there have been breakthroughs with pathogens such as Streptococcus and Meningitis.[60] Methods of vaccine production, such as biochemical and serological, are laborious and unreliable.They require the pathogens to be in vitro to be effective.[61] New advances in genomic development help predict nearly all variations of pathogens, thus making advances for vaccines.[61] Protein-based vaccines are being developed to combat resistant pathogens such as Staphylococcus and Chlamydia.[60]
Countering bioterrorism
In 2005, the sequence of the 1918 Spanish influenza was completed. Accompanied with phylogenetic analysis, it was possible to supply a detailed account of the virus' evolution and behavior, in particular its adaptation to humans.[62] Following the sequencing of the Spanish influenza, the pathogen was also reconstructed. When inserted into mice, the pathogen proved to be incredibly deadly.[63][11] The 2001 anthrax attacks shed light on the possibility of bioterrorism as being more of a real than imagined threat. Bioterrorism was anticipated in the Iraq war, with soldiers being inoculated for a smallpox attack.[64] Using technologies and insight gained from reconstruction of the Spanish influenza, it may be possible to prevent future deadly planted outbreaks of disease. There is a strong ethical concern however, as to whether the resurrection of old viruses is necessary and whether it does more harm than good.[11][65] The best avenue for countering such threats is coordinating with organizations which provide immunizations. The increased awareness and participation would greatly decrease the effectiveness of a potential epidemic. An addition to this measure would be to monitor natural water reservoirs as a basis to prevent an attack or outbreak. Overall, communication between labs and large organizations, such as Global Outbreak Alert and Response Network (GOARN), can lead to early detection and prevent outbreaks.[59]
See also
References
- Sharma AK, Dhasmana N, Dubey N, Kumar N, Gangwal A, Gupta M, Singh Y (March 2017). "Bacterial Virulence Factors: Secreted for Survival". Indian Journal of Microbiology. 57 (1): 1–10. doi:10.1007/s12088-016-0625-1. PMC 5243249. PMID 28148975.
- "How Pathogens Cause Disease | Microbiology". courses.lumenlearning.com. Retrieved 4 November 2019.
- Yang J, Chen L, Sun L, Yu J, Jin Q (January 2008). "VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics". Nucleic Acids Research. 36 (Database issue): D539-42. doi:10.1093/nar/gkm951. PMC 2238871. PMID 17984080.
- Gwinn M, MacCannell D, Armstrong GL (March 2019). "Next-Generation Sequencing of Infectious Pathogens". JAMA. 321 (9): 893–894. doi:10.1001/jama.2018.21669. PMC 6682455. PMID 30763433.
- Threats, Institute of Medicine (US) Forum on Microbial (2013). Workshop Overview. National Academies Press (US). Retrieved 8 November 2019.
- Ekundayo TC, Okoh AI (2018). "Plesiomonas shigelloides That Were Deemed Inconclusive by Traditional Experimental Approaches". Frontiers in Microbiology. 9: 3077. doi:10.3389/fmicb.2018.03077. PMC 6309461. PMID 30627119.
- Threats, Institute of Medicine (US) Forum on Microbial (2013). Workshop Overview. National Academies Press (US). Retrieved 8 November 2019.
- Lynch T, Petkau A, Knox N, Graham M, Van Domselaar G (October 2016). "A Primer on Infectious Disease Bacterial Genomics". Clinical Microbiology Reviews. 29 (4): 881–913. doi:10.1128/CMR.00001-16. PMC 5010755. PMID 28590251.
- Demuth A, Aharonowitz Y, Bachmann TT, Blum-Oehler G, Buchrieser C, Covacci A, et al. (May 2008). "Pathogenomics: an updated European Research Agenda". Infection, Genetics and Evolution. 8 (3): 386–93. doi:10.1016/j.meegid.2008.01.005. hdl:10033/30395. PMID 18321793.
- Vinatzer BA, Heath LS, Almohri HM, Stulberg MJ, Lowe C, Li S (15 May 2019). "Cyberbiosecurity Challenges of Pathogen Genome Databases". Frontiers in Bioengineering and Biotechnology. 7: 106. doi:10.3389/fbioe.2019.00106. PMC 6529814. PMID 31157218.
- Kaiser J (October 2005). "Virology. Resurrected influenza virus yields secrets of deadly 1918 pandemic". Science. 310 (5745): 28–9. doi:10.1126/science.310.5745.28. PMID 16210501. S2CID 26252589.
- Pallen MJ, Wren BW (October 2007). "Bacterial pathogenomics". Nature. 449 (7164): 835–42. Bibcode:2007Natur.449..835P. doi:10.1038/nature06248. PMID 17943120. S2CID 4313623.
- Brownlee GG (19 August 2015). "Frederick Sanger CBE CH OM. 13 August 1918 — 19 November 2013". Biographical Memoirs of Fellows of the Royal Society. 61: 437–466. doi:10.1098/rsbm.2015.0013.
- Willey JM (2020). Prescott's microbiology. New York, New York: McGraw-Hill Education. pp. 431–432. ISBN 9781260211887. OCLC 1039422993.
- "Timeline: Organisms that have had their genomes sequenced". Your Genome. 19 January 2015. Retrieved 9 November 2019.
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. (July 1995). "Whole-genome random sequencing and assembly of Haemophilus influenzae Rd". Science. 269 (5223): 496–512. Bibcode:1995Sci...269..496F. doi:10.1126/science.7542800. PMID 7542800.
- "Key Differences between next-generation sequencing and Sanger sequencing".
- Fraser-Liggett CM (December 2005). "Insights on biology and evolution from microbial genome sequencing". Genome Research. 15 (12): 1603–10. doi:10.1101/gr.3724205. PMID 16339357.
- Oakeson KF, Wagner JM, Mendenhall M, Rohrwasser A, Atkinson-Dunn R (September 2017). "Bioinformatic Analyses of Whole-Genome Sequence Data in a Public Health Laboratory". Emerging Infectious Diseases. 23 (9): 1441–1445. doi:10.3201/eid2309.170416. PMC 5572866. PMID 28820135.
- Lathe W, Williams J, Mangan M, Karolchik D (2008). "Genomic data resources: challenges and promises". Nature Education. p. 2.
- "VFDB: Virulence Factors of Bacterial Pathogens". www.mgc.ac.cn. Retrieved 8 November 2019.
- Rappuoli R (March 2001). "Reverse vaccinology, a genome-based approach to vaccine development". Vaccine. 19 (17–19): 2688–91. doi:10.1016/S0264-410X(00)00554-5. PMID 11257410.
- "Gene flow | genetics". Encyclopedia Britannica. Retrieved 4 November 2019.
- Griffiths AJ, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM (2000). "Sources of variation". An Introduction to Genetic Analysis (7th ed.). ISBN 978-0-7167-3771-1.
- "Comparative Genomics Fact Sheet". Genome.gov. Retrieved 13 November 2019.
- Hain T, Chatterjee SS, Ghai R, Kuenne CT, Billion A, Steinweg C, et al. (November 2007). "Pathogenomics of Listeria spp". International Journal of Medical Microbiology. 297 (7–8): 541–57. doi:10.1016/j.ijmm.2007.03.016. PMID 17482873.
- Perna NT, Plunkett G, Burland V, Mau B, Glasner JD, Rose DJ, et al. (January 2001). "Genome sequence of enterohaemorrhagic Escherichia coli O157:H7". Nature. 409 (6819): 529–33. Bibcode:2001Natur.409..529P. doi:10.1038/35054089. PMID 11206551.
- Koskiniemi S, Sun S, Berg OG, Andersson DI (June 2012). "Selection-driven gene loss in bacteria". PLOS Genetics. 8 (6): e1002787. doi:10.1371/journal.pgen.1002787. PMC 3386194. PMID 22761588.
- Bliven KA, Maurelli AT (December 2012). "Antivirulence genes: insights into pathogen evolution through gene loss". Infection and Immunity. 80 (12): 4061–70. doi:10.1128/iai.00740-12. PMC 3497401. PMID 23045475.
- Ward PN, Holden MT, Leigh JA, Lennard N, Bignell A, Barron A, et al. (January 2009). "Evidence for niche adaptation in the genome of the bovine pathogen Streptococcus uberis". BMC Genomics. 10: 54. doi:10.1186/1471-2164-10-54. PMC 2657157. PMID 19175920.
- Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, et al. (October 2001). "Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18". Nature. 413 (6858): 848–52. Bibcode:2001Natur.413..848P. doi:10.1038/35101607. PMID 11677608.
- Boucher Y, Douady CJ, Papke RT, Walsh DA, Boudreau ME, Nesbø CL, et al. (2003). "Lateral gene transfer and the origins of prokaryotic groups". Annual Review of Genetics. 37: 283–328. doi:10.1146/annurev.genet.37.050503.084247. PMID 14616063.
- Lima WC, Paquola AC, Varani AM, Van Sluys MA, Menck CF (April 2008). "Laterally transferred genomic islands in Xanthomonadales related to pathogenicity and primary metabolism". FEMS Microbiology Letters. 281 (1): 87–97. doi:10.1111/j.1574-6968.2008.01083.x. PMID 18318843.
- Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, et al. (April 2005). "Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain". Journal of Bacteriology. 187 (7): 2426–38. doi:10.1128/JB.187.7.2426-2438.2005. PMC 1065214. PMID 15774886.
- Bapteste E, Boucher Y (May 2008). "Lateral gene transfer challenges principles of microbial systematics". Trends in Microbiology. 16 (5): 200–7. doi:10.1016/j.tim.2008.02.005. PMID 18420414.
- Huang J, Gogarten JP (July 2006). "Ancient horizontal gene transfer can benefit phylogenetic reconstruction". Trends in Genetics. 22 (7): 361–6. doi:10.1016/j.tig.2006.05.004. PMID 16730850.
- Mira A, Ochman H, Moran NA (October 2001). "Deletional bias and the evolution of bacterial genomes". Trends in Genetics. 17 (10): 589–96. doi:10.1016/S0168-9525(01)02447-7. PMID 11585665.
- Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, Prentice MB, et al. (October 2001). "Genome sequence of Yersinia pestis, the causative agent of plague". Nature. 413 (6855): 523–7. Bibcode:2001Natur.413..523P. doi:10.1038/35097083. PMID 11586360.
- Nierman WC, DeShazer D, Kim HS, Tettelin H, Nelson KE, Feldblyum T, et al. (September 2004). "Structural flexibility in the Burkholderia mallei genome". Proceedings of the National Academy of Sciences of the United States of America. 101 (39): 14246–51. Bibcode:2004PNAS..10114246N. doi:10.1073/pnas.0403306101. PMC 521142. PMID 15377793.
- Holden MT, Titball RW, Peacock SJ, Cerdeño-Tárraga AM, Atkins T, Crossman LC, et al. (September 2004). "Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei". Proceedings of the National Academy of Sciences of the United States of America. 101 (39): 14240–5. doi:10.1073/pnas.0403302101. PMC 521101. PMID 15377794.
- "What are single nucleotide polymorphisms (SNPs)?". Genetics Home Reference. Retrieved 8 November 2019.
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. (September 2005). "Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome"". Proceedings of the National Academy of Sciences of the United States of America. 102 (39): 13950–5. Bibcode:2005PNAS..10213950T. doi:10.1073/pnas.0506758102. PMC 1216834. PMID 16172379.
- Lapierre P, Gogarten JP (March 2009). "Estimating the size of the bacterial pan-genome". Trends in Genetics. 25 (3): 107–10. doi:10.1016/j.tig.2008.12.004. PMID 19168257.
- Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R (December 2005). "The microbial pan-genome". Current Opinion in Genetics & Development. 15 (6): 589–94. doi:10.1016/j.gde.2005.09.006. PMID 16185861.
- Tettelin H, Riley D, Cattuto C, Medini D (October 2008). "Comparative genomics: the bacterial pan-genome". Current Opinion in Microbiology. 11 (5): 472–7. doi:10.1016/j.mib.2008.09.006. PMID 19086349.
- Gennari M, Ghidini V, Caburlotto G, Lleo MM (December 2012). "Virulence genes and pathogenicity islands in environmental Vibrio strains nonpathogenic to humans". FEMS Microbiology Ecology. 82 (3): 563–73. doi:10.1111/j.1574-6941.2012.01427.x. PMID 22676367.
- Langille MG, Brinkman FS (March 2009). "IslandViewer: an integrated interface for computational identification and visualization of genomic islands". Bioinformatics. 25 (5): 664–5. doi:10.1093/bioinformatics/btp030. PMC 2647836. PMID 19151094.
- Guy L (October 2006). "Identification and characterization of pathogenicity and other genomic islands using base composition analyses". Future Microbiology. 1 (3): 309–16. doi:10.2217/17460913.1.3.309. PMID 17661643.
- "Research on microbial biofilms (PA-03-047)". NIH, National Heart, Lung, and Blood Institute. 20 December 2002.
- Valle J, Vergara-Irigaray M, Merino N, Penadés JR, Lasa I (April 2007). "sigmaB regulates IS256-mediated Staphylococcus aureus biofilm phenotypic variation". Journal of Bacteriology. 189 (7): 2886–96. doi:10.1128/JB.01767-06. PMC 1855799. PMID 17277051.
- Hogardt M, Hoboth C, Schmoldt S, Henke C, Bader L, Heesemann J (January 2007). "Stage-specific adaptation of hypermutable Pseudomonas aeruginosa isolates during chronic pulmonary infection in patients with cystic fibrosis". The Journal of Infectious Diseases. 195 (1): 70–80. doi:10.1086/509821. PMID 17152010.
- Cheng LW, Viala JP, Stuurman N, Wiedemann U, Vale RD, Portnoy DA (September 2005). "Use of RNA interference in Drosophila S2 cells to identify host pathways controlling compartmentalization of an intracellular pathogen". Proceedings of the National Academy of Sciences of the United States of America. 102 (38): 13646–51. Bibcode:2005PNAS..10213646C. doi:10.1073/pnas.0506461102. PMC 1224656. PMID 16157870.
- Hattori M, Taylor TD (February 2009). "The human intestinal microbiome: a new frontier of human biology". DNA Research. 16 (1): 1–12. doi:10.1093/dnares/dsn033. PMC 2646358. PMID 19147530.
- Hooper LV, Gordon JI (May 2001). "Commensal host-bacterial relationships in the gut". Science. 292 (5519): 1115–8. Bibcode:2001Sci...292.1115H. doi:10.1126/science.1058709. PMID 11352068. S2CID 44645045.
- Oshima K, Toh H, Ogura Y, Sasamoto H, Morita H, Park SH, et al. (December 2008). "Complete genome sequence and comparative analysis of the wild-type commensal Escherichia coli strain SE11 isolated from a healthy adult". DNA Research. 15 (6): 375–86. doi:10.1093/dnares/dsn026. PMC 2608844. PMID 18931093.
- Zoetendal EG, Rajilic-Stojanovic M, de Vos WM (November 2008). "High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota". Gut. 57 (11): 1605–15. doi:10.1136/gut.2007.133603. PMID 18941009. S2CID 34347318.
- Achtman M, Morelli G, Zhu P, Wirth T, Diehl I, Kusecek B, et al. (December 2004). "Microevolution and history of the plague bacillus, Yersinia pestis". Proceedings of the National Academy of Sciences of the United States of America. 101 (51): 17837–42. Bibcode:2004PNAS..10117837A. doi:10.1073/pnas.0408026101. PMC 535704. PMID 15598742.
- Oyston PC, Dorrell N, Williams K, Li SR, Green M, Titball RW, Wren BW (June 2000). "The response regulator PhoP is important for survival under conditions of macrophage-induced stress and virulence in Yersinia pestis". Infection and Immunity. 68 (6): 3419–25. doi:10.1128/IAI.68.6.3419-3425.2000. PMC 97616. PMID 10816493.
- Pompe S, Simon J, Wiedemann PM, Tannert C (July 2005). "Future trends and challenges in pathogenomics. A Foresight study". EMBO Reports. 6 (7): 600–5. doi:10.1038/sj.embor.7400472. PMC 1369123. PMID 15995675.
- Sette A, Rappuoli R (October 2010). "Reverse vaccinology: developing vaccines in the era of genomics". Immunity. 33 (4): 530–41. doi:10.1016/j.immuni.2010.09.017. PMC 3320742. PMID 21029963.
- Rappuoli R (October 2000). "Reverse vaccinology". Current Opinion in Microbiology. 3 (5): 445–50. doi:10.1016/S1369-5274(00)00119-3. PMID 11050440.
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG (October 2005). "Characterization of the 1918 influenza virus polymerase genes". Nature. 437 (7060): 889–93. Bibcode:2005Natur.437..889T. doi:10.1038/nature04230. PMID 16208372. S2CID 4405787.
- Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solórzano A, Swayne DE, et al. (October 2005). "Characterization of the reconstructed 1918 Spanish influenza pandemic virus". Science. 310 (5745): 77–80. Bibcode:2005Sci...310...77T. CiteSeerX 10.1.1.418.9059. doi:10.1126/science.1119392. PMID 16210530. S2CID 14773861.
- "Terrorism Project)". Center for Defense Information. 20 December 2002.
- van Aken J (January 2007). "Ethics of reconstructing Spanish flu: is it wise to resurrect a deadly virus?". Heredity. 98 (1): 1–2. doi:10.1038/sj.hdy.6800911. PMID 17035950. S2CID 32686445.
