Pseudoachondroplasia
Pseudoachondroplasia is an inherited disorder of bone growth.[1] It is a genetic autosomal dominant disorder. It is generally not discovered until 2–3 years of age, since growth is normal at first. Pseudoachondroplasia is usually first detected by a drop of linear growth in contrast to peers, a waddling gait or arising lower limb deformities.[2]
| Pseudoachondroplasia | |
|---|---|
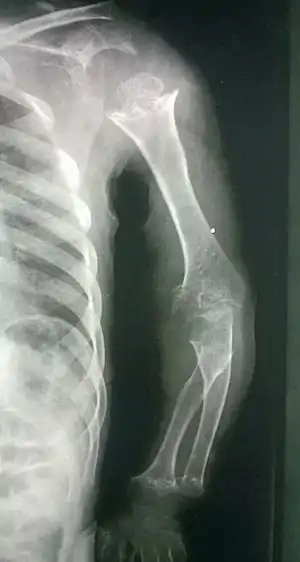 | |
| Pseudoachondroplasia. Shoulders and Humeri. Note the dysplastic proximal humeral epiphyses, metaphyseal broadening, irregularity and metaphyseal line of ossification. These changes are collectively known as "rachitic-like changes". Lesions are bilateral and symmetrical. | |
| Specialty | Medical genetics |
Pseudoachondroplasia (also known as PSACH, pseudoachondroplastic dysplasia, and pseudoachondroplastic spondyloepiphyseal dysplasia syndrome) is an osteochondrodysplasia that results in mild to severely short stature due to the inhibition of skeletal growth primarily in the limbs.[2] Though similarities in nomenclature may cause confusion, pseudoachondroplasia should not be confused with achondroplasia, which is a clinically and genetically distinct skeletal dysplasia. Pseudoachondroplasia is caused by a heterozygous mutation in the gene encoding cartilage oligomeric matrix protein (COMP). Mutation in the COMP gene can also cause multiple epiphyseal dysplasia. Despite the radioclinical similarities between pseudoachondroplasia and multiple epiphyseal dysplasia, the latter is less severe.[3]
Signs and symptoms
Disproportionate short stature, deformity of the lower limbs, short fingers, and ligamentous laxity give pseudoachondroplasia its distinctive features. The average height of adult males with the condition is around 120 centimeters (3 ft, 11 in), while adult females are typically around 116 cm (3 ft, 9in). Affected individuals are not noticeably short at birth. Patients with pseudoachondroplasia present with gait abnormalities, lower limb deformity, or a retarded growth rate that characteristically appear at age 2–3 years. Disproportionate short stature is characterized by shortening of proximal limb segments (humeri and femora) also called rhizomelic shortening. Other known clinical features include, genu valgum/varum, brachydactyly (short fingers), supple flexion deformity of the hips, knees, hyperlordosis of lumbar spine, rocker bottom feet and broadening of the metaphyseal ends of long bones especially around the wrists, knees and ankles. Patients with pseudoachondroplasia have normal intelligence and craniofacial features.[2][4][5]
 Pseudoachondroplasia
Pseudoachondroplasia Pseudoachondroplasia
Pseudoachondroplasia Pseudoachondroplasia
Pseudoachondroplasia
Genetics
Pseudoachondroplasia is inherited in an autosomal dominant manner, though one case of a very rare autosomal recessive form has been documented. The offspring of affected individuals are at 50% risk of inheriting the mutant allele. Prenatal testing by molecular genetic examination is available if the disease-causing mutation has been identified in an affected family member (Hecht et al. 1995).
Pathophysiology
COMP is an extracellular calcium binding protein directly involved in chondrocyte migration and proliferation. It is observed at a high frequency in chondrocytes in developing bone and tendon. In pseudochondroplasia, COMP is not secreted, but instead collects in the chondrocytes, ultimately poisoning and killing them. Though some chondrocytes do manage to survive, growth is significantly reduced, resulting in the characteristically short limbs and seemingly unaffected face and torso of those inflicted with the disorder (OMIM 2008). Mutations in COMP result in a phenotypic spectrum that varies from pseudochondroplasia (at the most extreme end) to multiple epiphyseal dysplasia or MED (a genetically similar, though milder skeletal dysplasia).[5]
Studies conducted by Hetch et al. suggest that type IX collagen, a collagen active specifically in the construction of cartilage, plays a key role in pseudoachondroplasia. The researchers found that IX collagen was amassed within the pseudoachondroplasia chondrocytes. This discovery suggests that the pathogenesis of pseudoachondroplasia involves the interactions of the mutant COMP gene products with specific cartilage components, such as type IX collagen, and that it is not solely the result of the effects of mutant molecules on the production and secretion of COMP (OMIM 2008).
Molecular biology
The COMP gene is located on chromosome 19p13.1; its precise locus is P49747. COMP contains 19 exons. The cartilage oligomeric matrix protein is 757 aa (OMIM 2008). COMP protein is found in the extracellular matrix, a complex web of proteins and other molecules that form in the spaces between the cells that make up ligaments and tendons. It is also found near chondrocytes (cartilage-forming cells). Chondrocytes play a vital role in osteogenesis (the formation of bone), particularly in the spine, hips, and limbs where osteogenesis begins with the formation of cartilage, which is then calcified and transformed into bone. We do not yet fully understand the normal function of COMP protein, though it is believed to play a part in cellular growth, division and apoptosis (self-destruction) of cells, as well as participating in the regulation of cell movement and attachment (OMIM 2008).
Nearly 60 mutations in the COMP gene have been identified in individuals with pseudoachondroplasia. However, the mutation responsible for the most common allele is a deletion of one codon within a very short triplet repeat (GAC), in which the 469th amino acid, an aspartic acid, is deleted (OMIM 2008).
Diagnosis
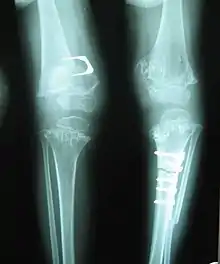
Exact diagnosis remains widely built on precise history taking, with the characteristic clinical and radiographic skeletal features.[2][4] Genetic diagnosis is based on DNA sequencing. Because plasma COMP levels are significantly reduced in patients with COMP mutations, such as pseudoachondroplasia, measuring plasma COMP levels has become a reliable means of diagnosing this and pathopysiologically similar disorders.[6]
Skeletal radiography
Accurate assessment of plain radiographic findings remains an important contributor to diagnosis of pseudoachondroplasia. It is noteworthy that vertebral radiographic abnormalities tend to resolve over time. Epiphyseal abnormalities tend to run a progressive course. Patients usually suffer early-onset arthritis of hips and knees. Many unique skeletal radiographic abnormalities of patients with pseudoachondroplasia have been reported in the literature.[2][7][4]
- Together with rhizomelic limb shortening, the presence of epiphyseal-metaphyseal changes of the long bones is a distinctive radiologic feature of pseudoachondroplasia.
- Hypoplastic capital femoral epiphyses, broad short femoral necks, coxa vara, horizontality of acetabular roof and delayed eruption of secondary ossification center of os pubis and greater trochanter.
- Dysplastic/hypoplastic epiphyses especially of shoulders and around the knees.
- Metaphyseal broadening, irregularity and metaphyseal line of ossification. These abnormalities that are typically encountered in proximal humerus and around the knees are collectively known as “rachitic-like changes”.
- Radiographic lesions of the appendicular skeleton are typically bilateral and symmetric.
- Oval shaped vertebrae with anterior beak originating and platyspondyly demonstrated on lateral radiographs of the spine.
- Normal widening of the interpedicular distances caudally demonstrated on anteroposterior radiographs of the dorsolumbar region. This is an important differentiating feature between pseudoachondroplasia and achondroplasia.
- Odontoid hypoplasia may occur resulting in cervical instability.
Differential diagnosis
- Achondroplasia[7]
- Multiple epiphyseal dysplasia[7]
- Mucopolysaccharidosis[7]
- Other causes of genu valgum (knock knees) or genu varum (bow legs) such as rickets
- Spondyloepiphyseal dysplasia congenita
- Radiographic findings of the pelvis and hips found in Perthes disease should not be confused with pseudochondroplasia. Patients with Perthes disease may present with unilateral hip affection. Besides bilateral hip affection are usually asymmetric. In contrast patients with pseudochondroplasia typically exhibit bilateral and symmetric hip involvement.[2][7]
Treatment
There is currently no cure for pseudoachondroplasia. However, management of the various health problems that result from the disorder includes medications such as analgesics (painkillers) for joint discomfort, osteotomy for lower limb deformities, and the surgical treatment of scoliosis. Prevention of some related health problems includes physical therapy to preserve joint flexibility and regular examinations to detect degenerative joint disease and neurological manifestations (particularly spinal cord compression). Additionally, healthcare providers recommend treatment for psychosocial issues related to short stature and other physical deformities for both affected individuals and their families (OMIM 2008).
Epidemiology
Pseudoachondroplasia is one of the most common skeletal dysplasias affecting all racial groups. However, no precise incidence figures are currently available (Suri et al. 2004).
History
In 1995 the gene responsible for Pseudoachondroplasia was identified by a research team led by Dr. Jacqueline Hecht of The University of Texas-Houston, Health Science Center. This discovery additionally shed light on the COMP protein, which the team recognized as somehow involved in skeletal growth and height determination (Hetch et al. 1995).[8]
In 1997, Hetch and her colleagues from the Research Department at Shriners Hospital for Children in Portland, Oregon conducted further research, which led to their discovery that the intracellular fate of mutant COMP is determined by the environment of individual chondrocytes, contrary to the previous notion that COMP activities leading to Pseudoachondroplasia were determined by structural effects of the mutation on COMP; this meant that COMP activities are cell-specific (Hetch et al. 1995).
Hetch et al. also discovered type IX collagen accumulated within the Pseudoachondroplasia chondrocytes. This discovery indicated the pathogenesis of Pseudoachondroplasia results from the interactions of the products of the mutant COMP allele with certain “cartilage components,” particularly with type IX collagen (Hetch et al. 1995).
See also
References
- Reference, Genetics Home. "pseudoachondroplasia". Genetics Home Reference. Retrieved 2017-09-27.
- Gamal R, Elsayed SM, EL-Sobky TA, Elabd HS (2017). "Pseudoachondroplasia in a child: the role of anthropometric measurements and skeletal imaging in differential diagnosis". The Egyptian Journal of Radiology and Nuclear Medicine. 48 (1): 245–50. doi:10.1016/j.ejrnm.2016.10.007.CS1 maint: multiple names: authors list (link)
- 132400
- Moosa S, Nishimura G (2013). "Pseudoachondroplasia: report on a South African family". South African Journal of Radiology. 17: 65–7. doi:10.7196/SAJR.767 (inactive 2021-01-03).CS1 maint: DOI inactive as of January 2021 (link)
- Jackson GC, Mittaz-Crettol L, Taylor JA, Mortier GR, Spranger J, Zabel B; et al. (2012). "Pseudoachondroplasia and multiple epiphyseal dysplasia: a 7-year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution". Human Mutation. 33 (1): 144–57. doi:10.1002/humu.21611. PMC 3272220. PMID 21922596.CS1 maint: multiple names: authors list (link)
- Tufan AC, Satiroglu-Tufan NL, Jackson GC, Semerci CN, Solak S, Yagci B (2007). "Serum or plasma cartilage oligomeric matrix protein concentration as a diagnostic marker in pseudoachondroplasia: differential diagnosis of a family". Eur J Hum Genet. 15 (10): 1023–1028. doi:10.1038/sj.ejhg.5201882. PMID 17579668. S2CID 74377.CS1 maint: multiple names: authors list (link)
- EL-Sobky, TA; Shawky, RM; Sakr, HM; Elsayed, SM; Elsayed, NS; Ragheb, SG; Gamal, R (15 November 2017). "A systematized approach to radiographic assessment of commonly seen genetic bone diseases in children: A pictorial review". J Musculoskelet Surg Res. 1 (2): 25. doi:10.4103/jmsr.jmsr_28_17. S2CID 79825711.
- Adelson, Betty M. (2011). Dwarfism: Medical and Psychosocial Aspects of Profound Short Stature. The Johns Hopkins University Press. p. 39. ISBN 978-0-8018-8121-3.
External links
| Classification | |
|---|---|
| External resources |