SNP annotation
Single nucleotide polymorphism annotation (SNP annotation) is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.[1]
| Classification | Bioinformatics |
|---|---|
| Subclassification | Single-nucleotide polymorphism |
| Type of tools used | Functional annotation tools |
| Other subjects related | Genome project, Genomics |
Introduction
Single nucleotide polymorphisms (SNPs) play an important role in genome wide association studies because they act as primary biomarkers. SNPs are currently the marker of choice due to their large numbers in virtually all populations of individuals. The location of these biomarkers can be tremendously important in terms of predicting functional significance, genetic mapping and population genetics.[3] Each SNP represents a nucleotide change between two individuals at a defined location. SNPs are the most common genetic variant found in all individual with one SNP every 100–300 bp in some species.[4] Since there is a massive number of SNPs on the genome, there is a clear need to prioritize SNPs according to their potential effect in order to expedite genotyping and analysis. [5]
Annotating large numbers of SNPs is a difficult and complex process, which need computational methods to handle such a large dataset. Many tools available have been developed for SNP annotation in different organisms: some of them are optimized for use with organisms densely sampled for SNPs (such as humans), but there are currently few tools available that are species non-specific or support non-model organism data. The majority of SNP annotation tools provide computationally predicted putative deleterious effects of SNPs. These tools examine whether a SNP resides in functional genomic regions such as exons, splice sites, or transcription regulatory sites, and predict the potential corresponding functional effects that the SNP may have using a variety of machine-learning approaches. But the tools and systems that prioritize functionally significant SNPs, suffer from few limitations: First, they examine the putative deleterious effects of SNPs with respect to a single biological function that provide only partial information about the functional significance of SNPs. Second, current systems classify SNPs into deleterious or neutral group.[6]
Many annotation algorithms focus on single nucleotide variants (SNVs), considered more rare than SNPs as defined by their minor allele frequency (MAF).[7][8] As a consequence, training data for the corresponding prediction methods may be different and hence one should be careful to select the appropriate tool for a specific purpose. For the purposes of this article, "SNP" will be used to mean both SNP and SNV, but readers should bear in mind the differences.
SNP annotation
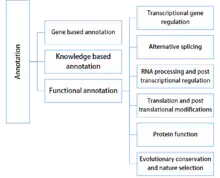
For SNP annotation, many kinds of genetic and genomic information are used. Based on the different features used by each annotation tool, SNP annotation methods may be split roughly into the following categories:
Gene based annotation
Genomic information from surrounding genomic elements is among the most useful information for interpreting the biological function of an observed variant. Information from a known gene is used as a reference to indicate whether the observed variant resides in or near a gene and if it has the potential to disrupt the protein sequence and its function. Gene based annotation is based on the fact that non-synonymous mutations can alter the protein sequence and that splice site mutation may disrupt the transcript splicing pattern.[9]
Knowledge based annotation
Knowledge base annotation is done based on the information of gene attribute, protein function and its metabolism. In this type of annotation more emphasis is given to genetic variation that disrupts the protein function domain, protein-protein interaction and biological pathway. The non-coding region of genome contain many important regulatory elements including promoter, enhancer and insulator, any kind of change in this regulatory region can change the functionality of that protein.[10] The mutation in DNA can change the RNA sequence and then influence the RNA secondary structure, RNA binding protein recognition and miRNA binding activity,.[11][12]
Functional annotation
This method mainly identifies variant function based on the information whether the variant loci are in the known functional region that harbor genomic or epigenomic signals. The function of non-coding variants are extensive in terms of the affected genomic region and they involve in almost all processes of gene regulation from transcriptional to post translational level [13]
Transcriptional gene regulation
Transcriptional gene regulation process depends on many spatial and temporal factors in the nucleus such as global or local chromatin states, nucleosome positioning, TF binding, enhancer/promoter activities. Variant that alter the function of any of these biological processes may alter the gene regulation and cause phenotypic abnormality.[14] Genetic variants that located in distal regulatory region can affect the binding motif of TFs, chromatin regulators and other distal transcriptional factors, which disturb the interaction between enhancer/silencer and its target gene.[15]
Alternative splicing
Alternative splicing is one of the most important components that show functional complexity of genome. Modified splicing has significant effect on the phenotype that is relevance to disease or drug metabolism. A change in splicing can be caused by modifying any of the components of the splicing machinery such as splice sites or splice enhancers or silencers.[16] Modification in the alternative splicing site can lead to a different protein form which will show a different function. Humans use an estimated 100,000 different proteins or more, so some genes must be capable of coding for a lot more than just one protein. Alternative splicing occurs more frequently than was previously thought and can be hard to control; genes may produce tens of thousands of different transcripts, necessitating a new gene model for each alternative splice.
RNA processing and post transcriptional regulation
Mutations in the untranslated region (UTR) affect many post-transcriptional regulation. Distinctive structural features are required for many RNA molecules and cis-acting regulatory elements to execute effective functions during gene regulation. SNVs can alter the secondary structure of RNA molecules and then disrupt the proper folding of RNAs, such as tRNA/mRNA/lncRNA folding and miRNA binding recognition regions.[17]
Translation and post translational modifications
Single nucleotide variant can also affect the cis-acting regulatory elements in mRNA’s to inhibit/promote the translation initiation. Change in the synonymous codons region due to mutation may affect the translation efficiency because of codon usage biases. The translation elongation can also be retarded by mutations along the ramp of ribosomal movement. In the post-translational level, genetic variants can contribute to proteostasis and amino acid modifications. However, mechanisms of variant effect in this field are complicated and there are only a few tools available to predict variant’s effect on translation related modifications.[18]
Protein function
Non-synonymous is the variant in exons that change the amino acid sequence encoded by the gene, including single base changes and non frameshift indels. It has been extremely investigated the function of non-synonymous variants on protein and many algorithms have been developed to predict the deleteriousness and pathogenesis of single nucleotide variants (SNVs). Classical bioinformatics tools, such as SIFT, Polyphen and MutationTaster, successfully predict the functional consequence of non-synonymous substitution.[19][20][21][22] PopViz webserver provides a gene-centric approach to visualize the mutation damage prediction scores (CADD, SIFT, PolyPhen-2) or the population genetics (minor allele frequency) versus the amino acid positions of all coding variants of a certain human gene.[23] PopViz is also cross-linked with UniProt database, where the protein domain information can be found, and to then identify the predicted deleterious variants fall into these protein domains on the PopViz plot.[23]
Evolutionary conservation and nature selection
Comparative genomics approaches were used to predict the function-relevant variants under the assumption that the functional genetic locus should be conserved across different species at an extensive phylogenetic distance. On the other hand, some adaptive traits and the population differences are driven by positive selections of advantageous variants, and these genetic mutations are functionally relevant to population specific phenotypes. Functional prediction of variants’ effect in different biological processes is pivotal to pinpoint the molecular mechanism of diseases/traits and direct the experimental validation.[24]
List of available SNP annotation tools
To annotate the vast amounts of available NGS data, currently a large number of SNPs annotation tools are available. Some of them are specific to specific SNPs while others are more general. Some of the available SNPs annotation tools are as follows SNPeff, Ensembl Variant Effect Predictor (VEP), ANNOVAR, FATHMM, PhD-SNP, PolyPhen-2, SuSPect, F-SNP, AnnTools, SeattleSeq, SNPit, SCAN, Snap, SNPs&GO, LS-SNP, Snat, TREAT, TRAMS, Maviant, MutationTaster, SNPdat, Snpranker, NGS – SNP, SVA, VARIANT, SIFT, LIST-S2, PhD-SNP and FAST-SNP. The functions and approaches used in SNPs annotation tools are listed below.
| Tools | Description | External resources use | WebsiteURL | References |
|---|---|---|---|---|
| PhyreRisk | Maps genetics variants onto experimental and predicted protein structures | Variant effect predictor, UniProt, Protein Data Bank, SIFTS, Phyre2 for predicted structures | http://phyrerisk.bc.ic.ac.uk/home | |
| Missense3D | Reports structural impact of a missense variant onto PDB and user-supplied protein coordinates. Developed to be applicable to experimental and predicted protein structures | Protein Data Bank, Phyre2 for predicted structures | http://www.sbg.bio.ic.ac.uk/~missense3d/ | |
| SNPeff | SnpEff annotates variants based on their genomic locations and predicts coding effects. Uses an interval forest approach | ENSEMBL, UCSC and organism based e.g. FlyBase, WormBase and TAIR | http://snpeff.sourceforge.net/SnpEff_manual.html | [27] |
| Ensembl VEP | Determines effects of variants (SNPs, insertions, deletions, CNVs or structural variants) on genes, transcripts, proteins and regulatory regions | dbSNP, RefSeq, UniProt, COSMIC, PDBe, 1000 Genomes, gnomAD, PubMed | https://www.ensembl.org/info/docs/tools/vep/index.html | [28] |
| ANNOVAR | This tool is suitable for pinpointing a small subset of functionally important variants. Uses mutation prediction approach for annotation | UCSC, RefSeq and Ensembl | http://annovar.openbioinformatics.org/ | [29] |
| Jannovar | This is a tool and library for genome annotation | RefSeq, Ensembl, UCSC, etc. | https://github.com/charite/jannovar | [30] |
| PhD-SNP | SVM-based method using sequence information retrieved by BLAST algorithm. | UniRef90 | http://snps.biofold.org/phd-snp/ | [31] |
| PolyPhen-2 | Suitable for predicting damaging effects of missense mutations. Uses sequence conservation, structure to model position of amino acid substitution, and SWISS-PROT annotation | UniProt | http://genetics.bwh.harvard.edu/pph2/ | [32] |
| MutationTaster | Suitable for predicting damaging effects of all intragenic mutations (DNA and protein level), including InDels. | Ensembl, 1000 Genomes Project, ExAC, UniProt, ClinVar, phyloP, phastCons, nnsplice, polyadq (...) | http://www.mutationtaster.org/ | [33] |
| SuSPect | An SVM-trained predictor of the damaging effects of missense mutations. Uses sequence conservation, structure and network (interactome) information to model phenotypic effect of amino acid substitution. Accepts VCF file | UniProt, PDB, Phyre2 for predicted structures, DOMINE and STRING for interactome | http://www.sbg.bio.ic.ac.uk/suspect/index.html | [34] |
| F-SNP | Computationally predicts functional SNPs for disease association studies. | PolyPhen, SIFT, SNPeffect, SNPs3D, LS-SNP, ESEfinder, RescueESE, ESRSearch, PESX, Ensembl, TFSearch, Consite, GoldenPath, Ensembl, KinasePhos, OGPET, Sulfinator, GoldenPath | http://compbio.cs.queensu.ca/F-SNP/ | [35] |
| AnnTools | Design to Identify novel and SNP/SNV, INDEL and SV/CNV. AnnTools searches for overlaps with regulatory elements, disease/trait associated loci, known segmental duplications and artifact prone regions | dbSNP, UCSC, GATK refGene, GAD, published lists of common structural genomic variation, Database of Genomic Variants, lists of conserved TFBs, miRNA | http://anntools.sourceforge.net/ | [36] |
| SNPit | Analyses the potential functional significance of SNPs derived from genome wide association studies | dbSNP, EntrezGene, UCSC Browser, HGMD, ECR Browser, Haplotter, SIFT | -/- | [37] |
| SCAN | Uses physical and functional based annotation to categorize according to their position relative to genes and according to linkage disequilibrium (LD) patterns and effects on expression levels | -/- | http://www.scandb.org/newinterface/about.html | [38] |
| SNAP | A neural network-based method for the prediction of the functional effects of non-synonymous SNPs | Ensembl, UCSC, Uniprot, UniProt, Pfam, DAS-CBS, MINT, BIND, KEGG, TreeFam | http://www.rostlab.org/services/SNAP | [39] |
| SNPs&GO | SVM-based method using sequence information, Gene Ontology annotation and when available protein structure. | UniRef90, GO, PANTHER, PDB | http://snps.biofold.org/snps-and-go/ | [40] |
| LS-SNP | Maps nsSNPs onto protein sequences, functional pathways and comparative protein structure models | UniProtKB, Genome Browser, dbSNP, PD | http://www.salilab.org/LS-SNP | [41] |
| TREAT | TREAT is a tool for facile navigation and mining of the variants from both targeted resequencing and whole exome sequencing | -/- | http://ndc.mayo.edu/mayo/research/biostat/stand-alone-packages.cfm | [42] |
| SNPdat | Suitable for species non-specific or support non-model organism data. SNPdat does not require the creation of any local relational databases or pre-processing of any mandatory input files | -/- | https://code.google.com/p/snpdat/downloads/ | [43] |
| NGS – SNP | Annotate SNPs comparing the reference amino acid and the non-reference amino acid to each orthologue | Ensembl, NCBI and UniProt | http://stothard.afns.ualberta.ca/downloads/NGS-SNP/ | [44] |
| SVA | Predicted biological function to variants identified | NCBI RefSeq, Ensembl, variation databases, UCSC, HGNC, GO, KEGG, HapMap, 1000 Genomes Project and DG | http://www.svaproject.org/ | [45] |
| VARIANT | VARIANT increases the information scope outside the coding regions by including all the available information on regulation, DNA structure, conservation, evolutionary pressures, etc. Regulatory variants constitute a recognized, but still unexplored, cause of pathologies | dbSNP,1000 genomes, disease-related variants from GWAS, OMIM, COSMIC | http://variant.bioinfo.cipf.es/ | [46] |
| SIFT | SIFT is a program that predicts whether an amino acid substitution affects protein function. SIFT uses sequence homology to predict whether an amino acid substitution will affect protein function | PROT/TrEMBL, or NCBI's | http://blocks.fhcrc.org/sift/SIFT.html | [47] |
| LIST-S2 | LIST-S2 (Local Identity and Shared Taxa, Species-specific) is based on the assumption that variations observed in closely related species are more significant when assessing conservation compared to those in distantly related species | UniProt SwissProt/TrEMBL and NCBI Taxonomy | https://gsponerlab.msl.ubc.ca/software/list/ | [48][49] |
| FAST-SNP | A web server that allows users to efficiently identify and prioritize high-risk SNPs according to their phenotypic risks and putative functional effects | NCBI dbSNP, Ensembl, TFSearch, PolyPhen, ESEfinder, RescueESE, FAS-ESS, SwissProt, UCSC Golden Path, NCBI Blast and HapMap | http://fastsnp.ibms.sinica.edu.tw/ | [50] |
| PANTHER | PANTHER relate protein sequence evolution to the evolution of specific protein functions and biological roles. The source of protein sequences used to build the protein family trees and used a computer-assisted manual curation step to better define the protein family clusters | STKE, KEGG, MetaCyc, FREX and Reactome | http://www.pantherdb.org/ | [51] |
| Meta-SNP | SVM-based meta predictor including 4 different methods. | PhD-SNP, PANTHER, SIFT, SNAP | http://snps.biofold.org/meta-snp | [52] |
| PopViz | Integrative and interactive gene-centric visualization of population genetics and mutation damage prediction scores of human gene variants | gnomAD, Ensembl, UniProt, OMIM, UCSC, CADD, EIGEN, LINSIGHT, SIFT, PolyPhen-2, | http://shiva.rockefeller.edu/PopViz/ | [23] |
Algorithms used in annotation tools
Variant annotation tools use machine learning algorithms to predict variant annotations. Different annotation tools use different algorithms. Common algorithms include:
- Interval/Random forest-eg.MutPred, SNPeff
- Neural networks-eg.SNAP
- Support Vector Machines-e.g. PhD-SNP, SNPs&GO
- Bayesian classification-eg.PolyPhen-2
Comparison of variant annotation tools
A large number of variant annotation tools are available for variant annotation. The annotation by different tools does not alway agree amongst each other, as the defined rules for data handling differ between applications. It is frankly impossible to perform a perfect comparison of the available tools. Not all tools have the same input and output nor the same functionality. Below is a table of major annotation tools and their functional area.
| Tools | Input file | Output file | SNP | INDEL | CNV | WEB or Program | Source | |
|---|---|---|---|---|---|---|---|---|
| AnnoVar | VCF, pileup,
CompleteGenomics, GFF3-SOLiD, SOAPsnp, MAQ, CASAVA |
TXT | Yes | Yes | Yes | Program | [53] | |
| Jannovar | VCF | VCF | Yes | Yes | Yes | Java Program | [54] | |
| SNPeff | VCF, pileup/TXT | VCF, TXT, HTML | Yes | Yes | No | Program | [55] | |
| Ensembl VEP | Ensembl default (coordinates), VCF, variant identifiers, HGVS, SPDI, REST-style regions | VCF, VEP, TXT, JSON | Yes | Yes | Yes | Web, Perl script, REST API | [56] | |
| AnnTools | VCF, pileup, TXT | VCF | Yes | Yes | No | No | [57] | |
| SeattleSeq | VVCF, MAQ, CASAVA,
GATK BED |
VCF, SeattleSeq | Yes | Yes | No | Web | [58] | |
| VARIANT | VCF, GFF2, BED | web report, TXT | Yes | Yes | Yes | Web | [59] |
Application
Different annotations capture diverse aspects of variant function.[61] Simultaneous use of multiple, varied functional annotations could improve rare variants association analysis power of whole exome and whole genome sequencing studies.[62]
Conclusions
The next generation of SNP annotation webservers can take advantage of the growing amount of data in core bioinformatics resources and use intelligent agents to fetch data from different sources as needed. From a user’s point of view, it is more efficient to submit a set of SNPs and receive results in a single step, which makes meta-servers the most attractive choice. However, if SNP annotation tools deliver heterogeneous data covering sequence, structure, regulation, pathways, etc., they must also provide frameworks for integrating data into a decision algorithm(s), and quantitative confidence measures so users can assess which data are relevant and which are not.
References
- Aubourg S, Rouzé P (2001). "Genome annotation". Plant Physiol. Biochem. 29 (3–4): 181–193. doi:10.1016/S0981-9428(01)01242-6.
- Karchin R (January 2009). "Next generation tools for the annotation of human SNPs". Briefings in Bioinformatics. 10 (1): 35–52. doi:10.1093/bib/bbn047. PMC 2638621. PMID 19181721.
- Shen TH, Carlson CS, Tarczy-Hornoch P (August 2009). "SNPit: a federated data integration system for the purpose of functional SNP annotation". Computer Methods and Programs in Biomedicine. 95 (2): 181–9. doi:10.1016/j.cmpb.2009.02.010. PMC 2680224. PMID 19327864.
- N. C. Oraguzie, E.H.A. Rikkerink, S.E. Gardiner, H.N. de Silva (eds.), "Association Mapping in Plants", Springer, 2007
- Capriotti E, Nehrt NL, Kann MG, Bromberg Y (July 2012). "Bioinformatics for personal genome interpretation". Briefings in Bioinformatics. 13 (4): 495–512. doi:10.1093/bib/bbr070. PMC 3404395. PMID 22247263.
- P. H. Lee, H. Shatkay, “Ranking single nucleotide polymorphisms by potential deleterious effects”, Computational Biology and Machine Learning Lab, School of Computing, Queen’s University, Kingston, ON, Canada
- "Single-nucleotide polymorphism", Wikipedia, 2019-08-12, retrieved 2019-09-03
- "Minor allele frequency", Wikipedia, 2019-08-12, retrieved 2019-09-03
- M. J. Li, J. Wang, "Current trend of annotating single nucleotide variation in humans – A case study on SNVrap", Elsevier, 2014, pp. 1–9
- Wang Z, Gerstein M, Snyder M (January 2009). "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews Genetics. 10 (1): 57–63. doi:10.1038/nrg2484. PMC 2949280. PMID 19015660.
- Halvorsen M, Martin JS, Broadaway S, Laederach A (August 2010). "Disease-associated mutations that alter the RNA structural ensemble". PLOS Genetics. 6 (8): e1001074. doi:10.1371/journal.pgen.1001074. PMC 2924325. PMID 20808897.
- Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, Chang HY (January 2014). "Landscape and variation of RNA secondary structure across the human transcriptome". Nature. 505 (7485): 706–9. Bibcode:2014Natur.505..706W. doi:10.1038/nature12946. PMC 3973747. PMID 24476892.
- Sauna ZE, Kimchi-Sarfaty C (August 2011). "Understanding the contribution of synonymous mutations to human disease". Nature Reviews Genetics. 12 (10): 683–91. doi:10.1038/nrg3051. PMID 21878961. S2CID 8358824.
- Li MJ, Yan B, Sham PC, Wang J (May 2015). "Exploring the function of genetic variants in the non-coding genomic regions: approaches for identifying human regulatory variants affecting gene expression". Briefings in Bioinformatics. 16 (3): 393–412. doi:10.1093/bib/bbu018. PMID 24916300.
- French JD, Ghoussaini M, Edwards SL, Meyer KB, Michailidou K, Ahmed S, et al. (April 2013). "Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers". American Journal of Human Genetics. 92 (4): 489–503. doi:10.1016/j.ajhg.2013.01.002. PMC 3617380. PMID 23540573.
- Faber K, Glatting KH, Mueller PJ, Risch A, Hotz-Wagenblatt A (2011). "Genome-wide prediction of splice-modifying SNPs in human genes using a new analysis pipeline called AASsites". BMC Bioinformatics. 12 Suppl 4 (Suppl 4): S2. doi:10.1186/1471-2105-12-s4-s2. PMC 3194194. PMID 21992029.
- Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, Almeida R, Zhernakova A, Reinmaa E, Võsa U, Hofker MH, Fehrmann RS, Fu J, Withoff S, Metspalu A, Franke L, Wijmenga C (2013). "Human disease-associated genetic variation impacts large intergenic non-coding RNA expression". PLOS Genetics. 9 (1): e1003201. doi:10.1371/journal.pgen.1003201. PMC 3547830. PMID 23341781.
- M. J. Li, J. Wang, "Current trend of annotating single nucleotide variation in humans – A case study on SNVrap", Elsevier, 2014, pp. 1–9
- J. Wu, R. Jiang, "Prediction of Deleterious Nonsynonymous Single-Nucleotide Polymorphism for Human Diseases", The Scientific World Journal, 2013, 10 pages
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (July 2012). "SIFT web server: predicting effects of amino acid substitutions on proteins". Nucleic Acids Research. 40 (Web Server issue): W452–7. doi:10.1093/nar/gks539. PMC 3394338. PMID 22689647.
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (April 2010). "A method and server for predicting damaging missense mutations". Nature Methods. 7 (4): 248–9. doi:10.1038/nmeth0410-248. PMC 2855889. PMID 20354512.
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D (August 2010). "MutationTaster evaluates disease-causing potential of sequence alterations". Nature Methods. 7 (8): 575–6. doi:10.1038/nmeth0810-575. PMID 20676075. S2CID 26892938.
- Zhang P, Bigio B, Rapaport F, Zhang S, Casanova J, Abel L, Boisson B, Itan Y, Stegle O (2018). "PopViz: a webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations". Bioinformatics. 34 (24): 4307–4309. doi:10.1093/bioinformatics/bty536. PMC 6289133. PMID 30535305.
- M. J. Li, J. Wang, "Current trend of annotating single nucleotide variation in humans – A case study on SNVrap", Elsevier, 2014, pp. 1–9
- Ofoegbu TC, David A, Kelley LA, Mezulis S, Islam SA, Mersmann SF, Stromich L, Vakser IA, Houlston RS, Sternberg MJ (2019). "PhyreRisk: A Dynamic Web Application to Bridge Genomics, Proteomics and 3D Structural Data to Guide Interpretation of Human Genetic Variants". J Mol Biol. 431 (13): 2460–2466. doi:10.1016/j.jmb.2019.04.043. PMC 6597944. PMID 31075275.
- Ittisoponpisan S, Islam SA, Khanna T, Alhuzimi E, David A, Sternberg MJ (2019). "Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated?". J Mol Biol. 431 (11): 2197–2212. doi:10.1016/j.jmb.2019.04.009. PMC 6544567. PMID 30995449.
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012). "A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3". Fly. 6 (2): 80–92. doi:10.4161/fly.19695. PMC 3679285. PMID 22728672.
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (June 2016). "The Ensembl Variant Effect Predictor". Genome Biology. 17 (1): 122. doi:10.1186/s13059-016-0974-4. PMC 4893825. PMID 27268795.
- Wang K, Li M, Hakonarson H (September 2010). "ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data". Nucleic Acids Research. 38 (16): e164. doi:10.1093/nar/gkq603. PMC 2938201. PMID 20601685.
- Jäger M, Wang K, Bauer S, Smedley D, Krawitz P, Robinson PN (May 2014). "Jannovar: a java library for exome annotation". Human Mutation. 35 (5): 548–55. doi:10.1002/humu.22531. PMID 24677618. S2CID 10822001.
- Capriotti E, Calabrese R, Casadio R (November 2006). "Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information" (PDF). Bioinformatics. 22 (22): 2729–34. doi:10.1093/bioinformatics/btl423. PMID 16895930.
- Adzhubei I, Jordan DM, Sunyaev SR (January 2013). "Predicting functional effect of human missense mutations using PolyPhen-2". Current Protocols in Human Genetics. Chapter 7: 7.20.1–7.20.41. doi:10.1002/0471142905.hg0720s76. PMC 4480630. PMID 23315928.
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D (August 2010). "MutationTaster evaluates disease-causing potential of sequence alterations". Nature Methods. 7 (8): 575–6. doi:10.1038/nmeth0810-575. PMID 20676075. S2CID 26892938.
- Yates CM, Filippis I, Kelley LA, Sternberg MJ (July 2014). "SuSPect: enhanced prediction of single amino acid variant (SAV) phenotype using network features". Journal of Molecular Biology. 426 (14): 2692–701. doi:10.1016/j.jmb.2014.04.026. PMC 4087249. PMID 24810707.
- Lee PH, Shatkay H (January 2008). "F-SNP: computationally predicted functional SNPs for disease association studies". Nucleic Acids Research. 36 (Database issue): D820–4. doi:10.1093/nar/gkm904. PMC 2238878. PMID 17986460.
- Makarov V, O'Grady T, Cai G, Lihm J, Buxbaum JD, Yoon S (March 2012). "AnnTools: a comprehensive and versatile annotation toolkit for genomic variants". Bioinformatics. 28 (5): 724–5. doi:10.1093/bioinformatics/bts032. PMC 3289923. PMID 22257670.
- Shen TH, Carlson CS, Tarczy-Hornoch P (August 2009). "SNPit: a federated data integration system for the purpose of functional SNP annotation". Computer Methods and Programs in Biomedicine. 95 (2): 181–9. doi:10.1016/j.cmpb.2009.02.010. PMC 2680224. PMID 19327864.
- Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, Dolan ME, Cox NJ (January 2010). "SCAN: SNP and copy number annotation". Bioinformatics. 26 (2): 259–62. doi:10.1093/bioinformatics/btp644. PMC 2852202. PMID 19933162.
- Bromberg Y, Rost B (2007). "SNAP: predict effect of non-synonymous polymorphisms on function". Nucleic Acids Research. 35 (11): 3823–35. doi:10.1093/nar/gkm238. PMC 1920242. PMID 17526529.
- Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R (August 2009). "Functional annotations improve the predictive score of human disease-related mutations in proteins" (PDF). Human Mutation. 30 (8): 1237–44. doi:10.1002/humu.21047. PMID 19514061. S2CID 33900765.
- Karchin R, Diekhans M, Kelly L, Thomas DJ, Pieper U, Eswar N, Haussler D, Sali A (June 2005). "LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources". Bioinformatics. 21 (12): 2814–20. doi:10.1093/bioinformatics/bti442. PMID 15827081.
- Asmann YW, Middha S, Hossain A, Baheti S, Li Y, Chai HS, Sun Z, Duffy PH, Hadad AA, Nair A, Liu X, Zhang Y, Klee EW, Kalari KR, Kocher JP (January 2012). "TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data". Bioinformatics. 28 (2): 277–8. doi:10.1093/bioinformatics/btr612. PMC 3259432. PMID 22088845.
- Doran AG, Creevey CJ (February 2013). "Snpdat: easy and rapid annotation of results from de novo snp discovery projects for model and non-model organisms". BMC Bioinformatics. 14: 45. doi:10.1186/1471-2105-14-45. PMC 3574845. PMID 23390980.
- Grant JR, Arantes AS, Liao X, Stothard P (August 2011). "In-depth annotation of SNPs arising from resequencing projects using NGS-SNP". Bioinformatics. 27 (16): 2300–1. doi:10.1093/bioinformatics/btr372. PMC 3150039. PMID 21697123.
- Ge D, Ruzzo EK, Shianna KV, He M, Pelak K, Heinzen EL, Need AC, Cirulli ET, Maia JM, Dickson SP, Zhu M, Singh A, Allen AS, Goldstein DB (July 2011). "SVA: software for annotating and visualizing sequenced human genomes". Bioinformatics. 27 (14): 1998–2000. doi:10.1093/bioinformatics/btr317. PMC 3129530. PMID 21624899.
- Medina I, De Maria A, Bleda M, Salavert F, Alonso R, Gonzalez CY, Dopazo J (July 2012). "VARIANT: Command Line, Web service and Web interface for fast and accurate functional characterization of variants found by Next-Generation Sequencing". Nucleic Acids Research. 40 (Web Server issue): W54–8. doi:10.1093/nar/gks572. PMC 3394276. PMID 22693211.
- Ng P. C.; Henikoff S. (2003). "SIFT: predicting amino acid changes that affect protein function". Nucleic Acids Research. 31 (13): 3812–3814. doi:10.1093/nar/gkg509. PMC 168916. PMID 12824425.
- Nawar Malhis; Steven J. M. Jones; Jörg Gsponer (2019). "Improved measures for evolutionary conservation that exploit taxonomy distances". Nature Communications. 10 (1): 1556. Bibcode:2019NatCo..10.1556M. doi:10.1038/s41467-019-09583-2. PMC 6450959. PMID 30952844.
- Nawar Malhis; Matthew Jacobson; Steven J. M. Jones; Jörg Gsponer (2020). "LIST-S2: Taxonomy Based Sorting of Deleterious Missense Mutations Across Species". Nucleic Acids Research. 48 (W1): W154–W161. doi:10.1093/nar/gkaa288. PMC 7319545. PMID 32352516.
- Yuan HY, Chiou JJ, Tseng WH, Liu CH, Liu CK, Lin YJ, Wang HH, Yao A, Chen YT, Hsu CN (July 2006). "FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization". Nucleic Acids Research. 34 (Web Server issue): W635–41. doi:10.1093/nar/gkl236. PMC 1538865. PMID 16845089.
- Mi H, Guo N, Kejariwal A, Thomas PD (January 2007). "PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways". Nucleic Acids Research. 35 (Database issue): D247–52. doi:10.1093/nar/gkl869. PMC 1716723. PMID 17130144.
- Capriotti E, Altman RB, Bromberg Y (2013). "Collective judgment predicts disease-associated single nucleotide variants". BMC Genomics. 14 Suppl 3: S2. doi:10.1186/1471-2164-14-S3-S2. PMC 3839641. PMID 23819846.
- Wang K, Li M, Hakonarson H (September 2010). "ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data". Nucleic Acids Research. 38 (16): e164. doi:10.1093/nar/gkq603. PMC 2938201. PMID 20601685.
- "charite/jannovar". GitHub. Retrieved 2016-09-25.
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012). "A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3". Fly. 6 (2): 80–92. doi:10.4161/fly.19695. PMC 3679285. PMID 22728672.
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (June 2016). "The Ensembl Variant Effect Predictor". Genome Biology. 17 (1): 122. doi:10.1186/s13059-016-0974-4. PMC 4893825. PMID 27268795.
- Makarov V, O'Grady T, Cai G, Lihm J, Buxbaum JD, Yoon S (March 2012). "AnnTools: a comprehensive and versatile annotation toolkit for genomic variants". Bioinformatics. 28 (5): 724–5. doi:10.1093/bioinformatics/bts032. PMC 3289923. PMID 22257670.
- "Input Variation List File for Annotation". SeattleSeq Annotation 151.
- Medina I, De Maria A, Bleda M, Salavert F, Alonso R, Gonzalez CY, Dopazo J (July 2012). "VARIANT: Command Line, Web service and Web interface for fast and accurate functional characterization of variants found by Next-Generation Sequencing". Nucleic Acids Research. 40 (Web Server issue): W54–8. doi:10.1093/nar/gks572. PMC 3394276. PMID 22693211.
- Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J, Trajanoski Z (March 2014). "A survey of tools for variant analysis of next-generation genome sequencing data". Briefings in Bioinformatics. 15 (2): 256–78. doi:10.1093/bib/bbs086. PMC 3956068. PMID 23341494.
- Lee, Phil H.; Lee, Christian; Li, Xihao; Wee, Brian; Dwivedi, Tushar; Daly, Mark (Jan 2018). "Principles and methods of in-silico prioritization of non-coding regulatory variants". Human Genetics. 137 (1): 15–30. doi:10.1007/s00439-017-1861-0. PMC 5892192. PMID 29288389.
- Li, Xihao; Li, Zilin; Zhou, Hufeng; Gaynor, Sheila M.; Liu, Yaowu; Chen, Han; Sun, Ryan; Dey, Rounak; Arnett, Donna K.; Aslibekyan, Stella; Ballantyne, Christie M.; Bielak, Lawrence F.; Blangero, John; Boerwinkle, Eric; Bowden, Donald W.; Broome, Jai G; Conomos, Matthew P; Correa, Adolfo; Cupples, L. Adrienne; Curran, Joanne E.; Freedman, Barry I.; Guo, Xiuqing; Hindy, George; Irvin, Marguerite R.; Kardia, Sharon L. R.; Kathiresan, Sekar; Khan, Alyna T.; Kooperberg, Charles L.; Laurie, Cathy C.; Liu, X. Shirley; Mahaney, Michael C.; Manichaiku, Ani W.; Martin, Lisa W.; Mathias, Rasika A.; McGarvey, Stephen T.; Mitchell, Braxton D.; Montasser, May E.; Moore, Jill E.; Morrison3, Alanna C.; O’Connell, Jeffrey R.; Palmer, Nicholette D.; Pampana, Akhil; Peralta, Juan M.; Peyser, Patricia A.; Psaty, Bruce M.; Redline, Susan; Rice, Kenneth M.; Rich, Stephen S.; Smith, Jennifer A.; Tiwari, Hemant K.; Tsai, Michael Y.; Vasan, Ramachandran S.; Wang, Fei Fei; Weeks, Daniel E.; Weng, Zhiping; Wilson, James G.; Yanek, Lisa R.; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; TOPMed Lipids Working Group; Neale, Benjamin M.; Sunyaev, Shamil R.; Abecasis, Gonçalo R.; Rotter, Jerome I.; Willer, Cristen J.; Peloso, Gina M.; Natarajan, Pradeep; Lin, Xihong (Sep 2020). "Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale". Nature Genetics. 52 (9): 969–983. doi:10.1038/s41588-020-0676-4. ISSN 1061-4036. PMC 7483769. PMID 32839606.
