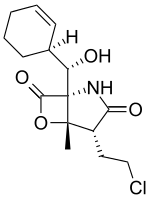
Salinosporamide A
Salinosporamide A (Marizomib) is a potent proteasome inhibitor being studied as a potential anticancer agent. It entered phase I human clinical trials for the treatment of multiple myeloma, only three years after its discovery in 2003.[1][2] This marine natural product is produced by the obligate marine bacteria Salinispora tropica and Salinispora arenicola, which are found in ocean sediment. Salinosporamide A belongs to a family of compounds, known collectively as salinosporamides, which possess a densely functionalized γ-lactam-β-lactone bicyclic core.
 | |
| Names | |
|---|---|
| IUPAC name
(4R,5S)-4-(2-chloroethyl)-1-((1S)-cyclohex-2-enyl(hydroxy)methyl)-5-methyl-6-oxa-2-azabicyclo[3.2.0]heptane-3,7-dione | |
| Other names
Marizomib; NPI-0052 | |
| Identifiers | |
3D model (JSmol) |
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
| KEGG | |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C15H20ClNO4 | |
| Molar mass | 313.781 g/mol |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
History
Salinosporamide A was discovered by William Fenical and Paul Jensen from Scripps Institution of Oceanography in La Jolla, CA. In preliminary screening, a high percentage of the organic extracts of cultured Salinispora strains possessed antibiotic and anticancer activities, which suggests that these bacteria are an excellent resource for drug discovery. Salinispora strain CNB-392 was isolated from a heat-treated marine sediment sample and cytotoxicity-guided fractionation of the crude extract led to the isolation of salinosporamide A. Although salinosporamide A shares an identical bicyclic ring structure with omuralide, it is uniquely functionalized. Salinosporamide A displayed potent in vitro cytotoxicity against HCT-116 human colon carcinoma with an IC50 value of 11 ng mL-1. This compound also displayed potent and highly selective activity in the NCI's 60-cell-line panel with a mean GI50 value (the concentration required to achieve 50% growth inhibition) of less than 10 nM and a greater than 4 log LC50 differential between resistant and susceptible cell lines. The greatest potency was observed against NCI-H226 non-small cell lung cancer, SF-539 brain tumor, SK-MEL-28 melanoma, and MDA-MB-435 melanoma (formerly misclassified as breast cancer[3]), all with LC50 values less than 10 nM. Salinosporamide A was tested for its effects on proteasome function because of its structural relationship to omuralide. When tested against purified 20S proteasome, salinosporamide A inhibited proteasomal chymotrypsin-like proteolytic activity with an IC50 value of 1.3 nM.[4] This compound is approximately 35 times more potent than omuralide which was tested as a positive control in the same assay. Thus, the unique functionalization of the core bicyclic ring structure of salinosporamide A appears to have resulted in a molecule that is a significantly more potent proteasome inhibitor than omuralide.[1]
Mechanism of action
Salinosporamide A inhibits proteasome activity by covalently modifying the active site threonine residues of the 20S proteasome.
Biosynthesis
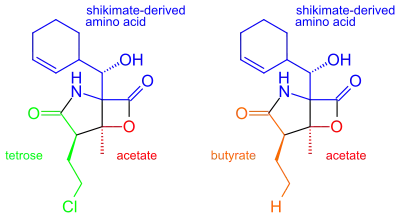
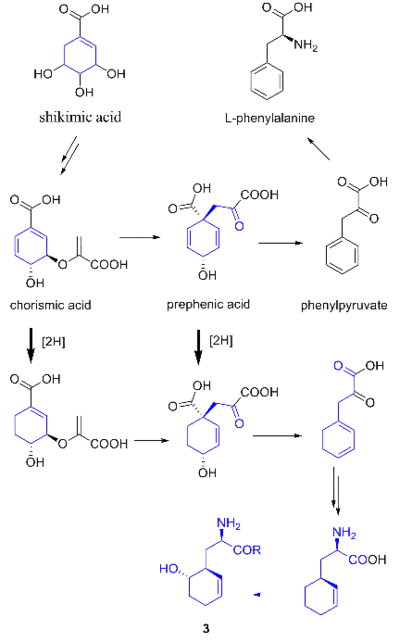
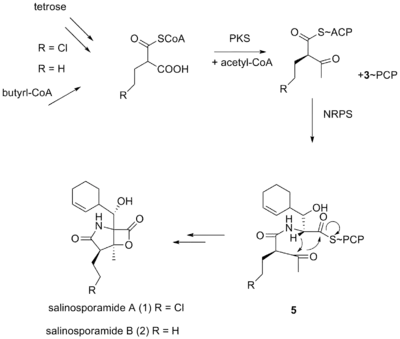
It was originally hypothesized that salinosporamide B was a biosynthetic precursor to salinosporamide A due to their structural similarities.
It was thought that the halogenation of the unactivated methyl group was catalyzed by a non-heme iron halogenase.[5][6] Recent work using 13C-labeled feeding experiments reveal distinct biosynthetic origins of salinosporamide A and B.[5][7]
While they share the biosynthetic precursors acetate and presumed β-hydroxycyclohex-2'-enylalanine (3), they differ in the origin of the four-carbon building block that gives rise to their structural differences involving the halogen atom. A hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) pathway is most likely the biosynthetic mechanism in which acetyl-CoA and butyrate-derived ethylmalonyl-CoA condense to yield the β-ketothioester (4), which then reacts with (3) to generate the linear precursor (5).
Total synthesis
The first stereoselective synthesis was reported by Rajender Reddy Leleti and E. J.Corey.[8] Later several routes to the total synthesis of salinosporamide A have been reported.[8][9][10][11]
Clinical study
In vitro studies using purified 20S proteasomes showed that salinosporamide A has lower EC50 for trypsin-like (T-L) activity than does bortezomib. In vivo animal model studies show marked inhibition of T-L activity in response to salinosporamide A, whereas bortezomib enhances T-L proteasome activity.
Initial results from early-stage clinical trials of salinosporamide A in relapsed/refractory multiple myeloma patients were presented at the 2011 American Society of Hematology annual meeting.[12] Further early-stage trials of the drug in a number of different cancers are ongoing.[13]
See also
- Salinosporamide B
References
- Feling RH; Buchanan GO; Mincer TJ; Kauffman CA; Jensen PR; Fenical W (2003). "Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora". Angew. Chem. Int. Ed. Engl. 42 (3): 355–7. doi:10.1002/anie.200390115. PMID 12548698.
- Chauhan D, Catley L, Li G, et al. (2005). "A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib". Cancer Cell. 8 (5): 407–19. doi:10.1016/j.ccr.2005.10.013. PMID 16286248.
- "MDA-MB-435, and its derivation MDA-N, are Melanoma cell lines, not breast cancer cell lines". Developmental Therapeutics Program. National Cancer Institute. 8 May 2015. Retrieved 6 January 2018.
- K. Lloyd, S. Glaser, B. Miller, Nereus Pharmaceuticals Inc.
- Beer LL; Moore BS (2007). "Biosynthetic convergence of salinosporamides A and B in the marine actinomycete Salinispora tropica". Org. Lett. 9 (5): 845–8. doi:10.1021/ol063102o. PMID 17274624.
- Vaillancourt FH; Yeh E; Vosburg DA; Garneau-Tsodikova S; Walsh CT (2006). "Nature's inventory of halogenation catalysts: oxidative strategies predominate". Chem. Rev. 106 (8): 3364–78. doi:10.1021/cr050313i. PMID 16895332.
- Tsueng G; McArthur KA; Potts BC; Lam KS (2007). "Unique butyric acid incorporation patterns for salinosporamides A and B reveal distinct biosynthetic origins". Applied Microbiology and Biotechnology. 75 (5): 999–1005. doi:10.1007/s00253-007-0899-7. PMID 17340108. S2CID 8992755.
- Reddy LR; Saravanan P; Corey EJ (2004). "A simple stereocontrolled synthesis of salinosporamide A". J. Am. Chem. Soc. 126 (20): 6230–1. doi:10.1021/ja048613p. PMID 15149210.
- Ling T; Macherla VR; Manam RR; McArthur KA; Potts BC (2007). "Enantioselective Total Synthesis of (−)-Salinosporamide A (NPI-0052)". Org. Lett. 9 (12): 2289–92. doi:10.1021/ol0706051. PMID 17497868.
- Ma G; Nguyen H; Romo D (2007). "Concise Total Synthesis of (±)-Salinosporamide A, (±)-Cinnabaramide A, and Derivatives via a Bis-Cyclization Process: Implications for a Biosynthetic Pathway?". Org. Lett. 9 (11): 2143–6. doi:10.1021/ol070616u. PMC 2518687. PMID 17477539.
- Endo A; Danishefsky SJ (2005). "Total synthesis of salinosporamide A". J. Am. Chem. Soc. 127 (23): 8298–9. doi:10.1021/ja0522783. PMID 15941259.
- "Marizomib May Be Effective In Relapsed/Refractory Multiple Myeloma (ASH 2011)". The Myeloma Beacon. 2012-01-23. Retrieved 2012-06-10.
- ClinicalTrials.gov: Marizomib
External links
 Media related to Salinosporamides at Wikimedia Commons
Media related to Salinosporamides at Wikimedia Commons