Transition state
The transition state of a chemical reaction is a particular configuration along the reaction coordinate. It is defined as the state corresponding to the highest potential energy along this reaction coordinate.[1] It is often marked with the double dagger ‡ symbol.
As an example, the transition state shown below occurs during the SN2 reaction of bromoethane with a hydroxyl anion:
![]()
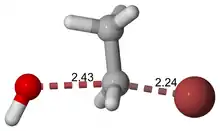
The activated complex of a reaction can refer to either the transition state or to other states along the reaction coordinate between reactants and products, especially those close to the transition state.[3]
According to the transition state theory, once the reactants have passed through the transition state configuration, they always continue to form products.[3]
History of concept
The concept of a transition state has been important in many theories of the rates at which chemical reactions occur. This started with the transition state theory (also referred to as the activated complex theory), which was first developed around 1935 by Eyring, Evans and Polanyi, and introduced basic concepts in chemical kinetics that are still used today.
Explanation
A collision between reactant molecules may or may not result in a successful reaction. The outcome depends on factors such as the relative kinetic energy, relative orientation and internal energy of the molecules. Even if the collision partners form an activated complex they are not bound to go on and form products, and instead the complex may fall apart back to the reactants.
Observing transition states
Because of the rules of quantum mechanics, the transition state cannot be captured or directly observed; the population at that point is zero. This is sometimes expressed by stating that the transition state has a fleeting existence. However, cleverly manipulated spectroscopic techniques can get us as close as the timescale of the technique allows. Femtochemical IR spectroscopy was developed for that reason, and it is possible to probe molecular structure extremely close to the transition point. Often, along the reaction coordinate, reactive intermediates are present not much lower in energy from a transition state making it difficult to distinguish between the two.
Determining the geometry of a transition state
Transition state structures can be determined by searching for first-order saddle points on the potential energy surface (PES) of the chemical species of interest.[4] A first-order saddle point is a critical point of index one, that is, a position on the PES corresponding to a minimum in all directions except one. This is further described in the article geometry optimization.
The Hammond–Leffler postulate
The Hammond–Leffler postulate states that the structure of the transition state more closely resembles either the products or the starting material, depending on which is higher in enthalpy. A transition state that resembles the reactants more than the products is said to be early, while a transition state that resembles the products more than the reactants is said to be late. Thus, the Hammond–Leffler Postulate predicts a late transition state for an endothermic reaction and an early transition state for an exothermic reaction.
A dimensionless reaction coordinate that quantifies the lateness of a transition state can be used to test the validity of the Hammond–Leffler postulate for a particular reaction.[5]
The structure–correlation principle
The structure–correlation principle states that structural changes that occur along the reaction coordinate can reveal themselves in the ground state as deviations of bond distances and angles from normal values along the reaction coordinate.[6] According to this theory if one particular bond length on reaching the transition state increases then this bond is already longer in its ground state compared to a compound not sharing this transition state. One demonstration of this principle is found in the two bicyclic compounds depicted below.[7] The one on the left is a bicyclo[2.2.2]octene, which, at 200 °C, extrudes ethylene in a retro-Diels–Alder reaction.

Compared to the compound on the right (which, lacking an alkene group, is unable to give this reaction) the bridgehead carbon-carbon bond length is expected to be shorter if the theory holds, because on approaching the transition state this bond gains double bond character. For these two compounds the prediction holds up based on X-ray crystallography.
Implications for enzymatic catalysis
One way that enzymatic catalysis proceeds is by stabilizing the transition state through electrostatics. By lowering the energy of the transition state, it allows a greater population of the starting material to attain the energy needed to overcome the transition energy and proceed to product.[8]
See also
- Transition state theory
- Transition state analogs, chemical compounds mimicking the substrate's transition state and act as enzyme inhibitors
- Reaction intermediate
- Reactive intermediate
- Activated complex
References
- Solomons, T.W. Graham & Fryhle, Craig B. (2004). Organic Chemistry (8th ed.). John Wiley & Sons, Inc. ISBN 0-471-41799-8.
- The calculation used a B3LYP functional and a 6-31+G* basis set.
- Peter Atkins and Julio de Paula, Physical Chemistry (8th ed., W.H. Freeman 2006), p.809 ISBN 0-7167-8759-8
- Frank Jensen (1999). Introduction to Computational Chemistry. England: John Wiley and Sons Ltd.
- Thomas A. Manz; David S. Sholl (2009). "A dimensionless reaction coordinate for quantifying the lateness of transition states". J. Comput. Chem.: NA. doi:10.1002/jcc.21440.
- Buergi, Hans Beat; Dunitz, Jack D. (1983). "From crystal statics to chemical dynamics". Accounts of Chemical Research. 16 (5): 153. doi:10.1021/ar00089a002.
- Goh, Yit Wooi; Danczak, Stephen M.; Lim, Tang Kuan; White, Jonathan M. (2007). "Manifestations of the Alder−Rickert Reaction in the Structures of Bicyclo[2.2.2]octadiene and Bicyclo[2.2.2]octene Derivatives". The Journal of Organic Chemistry. 72 (8): 2929–35. doi:10.1021/jo0625610. PMID 17371072.
- Srinivasan, Bharath (2020-09-27). "Words of advice: teaching enzyme kinetics". The FEBS Journal. doi:10.1111/febs.15537. ISSN 1742-464X.