Ube3a-ATS
UBE3A-ATS/Ube3a-ATS (human/mouse), otherwise known as ubiquitin ligase E3A-ATS, is the name for the antisense DNA strand that is transcribed as part of a larger transcript called LNCAT (large non-coding antisense transcript) at the Ube3a locus. The Ube3a locus is imprinted and in the central nervous system expressed only from the maternal allele. Silencing of Ube3a on the paternal allele is thought to occur through the Ube3a-ATS part of LNCAT,[1] since non-coding antisense transcripts are often found at imprinted loci.[2] The deletion and/or mutation of Ube3a on the maternal chromosome causes Angelman Syndrome (AS) and Ube3a-ATS may prove to be an important aspect in finding a therapy for this disease. While in patients with AS the maternal Ube3a allele is inactive, the paternal allele is intact but epigenetically silenced. If unsilenced, the paternal allele could be a source of active Ube3a protein in AS patients. Therefore, understanding the mechanisms of how Ube3a-ATS might be involved in silencing the paternal Ube3a may lead to new therapies for AS. This possibility has been demonstrated by a recent study where the drug topotecan, administered to mice suffering from AS, activated expression of the paternal Ube3a gene by lowering the transcription of Ube3a-ATS.[3]
LNCAT organization
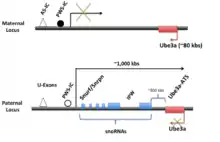
The human UBE3A-ATS is expressed as a part of LNCAT mainly from the paternal allele in the central nervous system (CNS).[1][6] The transcript is about 450 kbs long, starts at the U-exons and extends as far as UBE3A on the opposite strand, possibly beyond. The promoter for Snurf/Snrpn and the imprinting center are found in the U-exon region. The promoter region is imperative, as deletion of this area abolishes Ube3a-ATS transcription. Near the promoter is the PWS-IC and about 35 kbs upstream of the PWS-IC is the AS-IC. These two regions are thought to control the expression of the entire LNCAT strand. Starting at the promoter, the entire transcript can be transcribed and after transcription is further processed and spliced. Reviewed in Trends in Neurosci.[5]
Located next to the U-exon promoter region is Snrpn/Snurf which can be alternatively spliced into either Snrpn or Snurf in humans (in mice this remains as one bicistronic transcript).[7] Snrpn codes for a protein of unknown function which localizes to the cell nucleus. Snurf codes for a small nuclear ribonucleoprotein. While most of these proteins are involved in splicing, the role of this particular protein is not yet known.[7] Downstream from Snrpn/Snurf and within its introns are sequences for several C/D box snoRNAs. Most C/D box snoRNAs function in non-mRNA methylation.[7][8] However, recently, one snoRNA on Ube3a-ATS, SNORD 115, has been found to change the alternative splicing of the serotonin receptor 2C pre-mRNA. In addition, this snoRNA has the ability to change the splicing of five different mRNAs.[9] Among the sequences for the snoRNAs is nested IPW (imprinted Prader-Willi), a non-coding region whose deletion is thought to cause Prader-Willi syndrome.[10]
Model systems
The mouse and human Ube3a-ATS/Ube3a are orthologous and the general organizations of the regions are similar. For example, the mouse locus also contains Snurf/Snrpn, snoRNAs and IPW. The main differences are the locations and the lengths of the Ube3a-ATS transcripts. The human Ube3a/Ube3a-ATS is located on chromosome 15, while the mouse Ube3a is located on chromosome 7. The mouse LNCAT, including Ube3a-ATS, is about 1000 kb long, much longer than the human 450 kb LNCAT.[1][5][6] Due to the similar organization of the mouse and human LNCAT/Ube3a-ATS and the fact that the mouse Ube3a locus is also imprinted, the mouse is an excellent model system to study imprinting and the interactions between Ube3a/Ube3a-ATs. In addition, mouse neurons continue to retain their imprinting pattern in culture.[11]
Splice variants and locations
While the entire LNCAT transcript, including the Ube3a-ATS transcript may be transcribed, it is often spliced to include and exclude a variety of exons. Different splice variants are expressed in different tissue types and situations. For the most part, it is thought that at least some type of Ube3a-ATS is expressed in CNS cells that are imprinted, such as Purkinje cells and hippocampal neurons. However, there is spatiotemporal regulation of both the downstream and the upstream part of this transcript.[12] and Journal of Neuroscience.[13]
In mouse embryos, Snurf/Snrpn exons were detected in blastocysts about 7 days post coitem and continued to be expressed throughout development. The Snurf/Snrpn exons are restricted to CNS tissue during development, and only later during adulthood are expressed in other tissue. Ube3a-ATS exons were not detected until 10 days post coitem and their expression was also limited to the CNS during development. In general, Ube3a-ATS is detected during the initial stages of neurogenesis while Snurf/Snrpn is expressed in undifferentiated precursor cells and throughout the course of differentiation.[12] There are at least 10 different splice isoforms according to the UCSC genome browser.
According to one study, the splice variant that directly overlaps the Ube3a is found in the cytoplasm.[14]
Preventing expression on both alleles
A specific imprinting center cluster was thought to control the differential expression of Ube3a-ATS on the maternal and paternal alleles. There are two regions in the imprinting centers (ICs) that exist associated with AS and PWS- the AS-IC and the PWS-IC. These imprinting centers are control regions that dictate whether surrounding genes and regions can be expressed and are found on both the maternal and paternal alleles. While differential methylation patterns on the maternal and paternal genes are often associated with imprinting, the AS-IC remains unmethylated at both alleles. However, the neighboring PWS-IC is methylated on the maternal allele, but remains unmethylated on the paternal allele.[15]
The PWS-IC is suspected of controlling the expression of LNCAT and Ube3a-ATS. In mice where the PWS-IC has been deleted, expression of the Ube3a-ATS is decreased.[4] In the central neural system, Ube3a-ATS is preferentially expressed from the paternal allele where the PWS-IC is not methylated.[12] On the other hand, on the maternal allele, where the PWS-IC is methylated, Ube3a-ATS is not expressed, suggesting that the methylation of the PWS-IC somehow prevents Ube3a-ATS expression. This is supported by several studies where preventing methylation of the PWS-IC by knocking out methyl transferases in embryonic stem cells results in bialellic expression of Ube3a-ATS and silencing of Ube3a on the maternal allele.[9]
However, methylation is not the only process involved in preventing the expression of the maternal Ube3a-ATS. It is expected that the imprinting domains interact with other proteins, which contribute to the silencing of LNCAT and Ube3a-ATS on the maternal allele. For example, when MECP2 is knocked out, such as in Rett Syndrome patients, Ube3a-ATS is biallelically expressed, decreasing expression of Ube3a from the maternal allele.[9]
Collision model
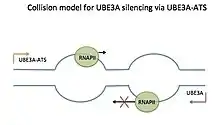
There are currently three models that explain how the Ube3a-ATS of LNCAT silences the paternal Ube3a- the collision model, the RNA-DNA interaction model, and the double stranded RNA interference model.[5][14] While these models have not been demonstrated directly for Ube3a/Ube3a-ATS, they are considered plausible based on evidence for the silencing of other natural antisense transcripts by these methods. However, the collision model, due to most recent supporting studies, appears most likely.[16][17][18]
The collision model can be thought of as a road wide enough for only one car. A smart car is traveling from one direction, and a plough from the other direction, eventually colliding. After the collision, the plough pushes the smart car backwards, as it continues to travel forward. In the collision model for Ube3a/Ube3a-ATS, RNA polymerases (RNAPs) travel towards each other along the sense and antisense templates during transcription. The sense and antisense templates overlap for Ube3a and Ube3a-ATS. The two transcription bubbles will collide head-on, and the RNAP transcribing the Ube3a-ATS, being the plough, will push the RNAP transcribing the Ube3a (smart car), backwards and eventually off of the template. This prevents complete transcription of Ube3a.[5]
The support for this model comes from two recent studies. The first study looked at transcription of genes on sense strands that were overlapped by genes being expressed on the antisense strand. The longer the region of overlap, the less efficient the transcription of the sense strand was, indicating that transcription on one strand interferes with the transcription on the other strand.[18] Another study directly monitored collisions between RNAPs transcribing a template using atomic force microscopy. RNAPs were stalled on DNA fragments and collided with other elongating RNAPs. The images showed stalling of the two RNAPs immediately after the collision, in addition to backtracking of one of the RNAPs.[16]
While these studies have not been performed for Ube3a/Ube3a-ATS, the use of atomic force microscopy to monitor transcription at this locus might provide insight as to how Ube3a is actually silenced via Ube3a-ATS. Further studies are still very much necessary to confirm these models for Ube3a.
Contradictory studies
While several studies support the idea that Ube3a-ATS might be involved in paternal Ube3a silencing, other studies contradict this. One study in particular argues against the in cis silencing of Ube3a by Ube3a-ATS. In this study, when the maternal Ube3a allele was deleted, an increase in paternal Ube3a-ATS expression was seen. This suggests that rather than the paternal Ube3a-ATS controlling paternal Ube3a, the maternal Ube3a somehow suppresses expression of the paternal Ube3a-ATs, possibly in trans rather than in cis. An interaction between the maternal and paternal homolgous regions of these genes was in fact observed in human and mouse cells during interphase.[14]
One mechanism to explain in trans silencing includes an interaction between the paternal Ube3a-ATS RNA and the maternal Ube3a mRNA. It is possible that the maternal Ube3a mRNA interacts with the paternal Ube3a-ATS RNA and decreases the stability of both of these transcripts. When only Ube3a-ATS is made without Ube3a, the Ube3a-ATS becomes more stable.[14]
Another study has suggested that Ube3a-ATS expression does not occur in imprinted regions. In situ hybridizations did not reveal Ube3a-ATS in Purkinje cells or hippocampal neurons. However, other upstream exons that correspond to Snurf/Snrpn were expressed,[12] indicating that the collision model could still be occurring. Thus further research is still required.
The future
Several studies have attempted to utilize the possibility of controlling Ube3a expression through Ube3a-ATS. In AS, the paternal PWS-IC is not methylated, supposedly allowing Ube3a-ATS expression. Therefore, if methylation of the PWS-IC were possible, Ube3a-ATS transcription could be prohibited, allowing Ube3a expression from the paternal allele to make up for the lack of expression from the maternal allele. A one year study was performed with several AS patients. These patients were put on methylation promoting diets that consisted of betaine, metafolin, creatine, and vitamin B12 supplements. However, after one year, methylation patterns in these patients did not change.[19]
Another study tested a large library of different drugs, and identified several topoisomerase I and II inhibitors which increased expression of paternal Ube3a in mouse neurons and mice. Topoisomerase inhibitors are widely used as chemotherapeutics and cause replicating cells to undergo apoptosis by inducing double strand breaks that stall the replication fork. However, their mechanism of action in activating the paternal Ube3a is not yet known, but may involve transcriptional interference with Ube3a-ATS, as Ube3a-ATS transcripts decreased after drug treatment. The group specifically chose to study topotecan, which was the most effective at a low nanomolar range and is already Food and Drug Administration approved for treating several types of cancers.[3]
References
- Runte, M.; Hüttenhofer, A; Gross, S; Kiefmann, M; Horsthemke, B; Buiting, K (2001). "The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A". Human Molecular Genetics. 10 (23): 2687–2700. doi:10.1093/hmg/10.23.2687. PMID 11726556.
- Royo, Hélène; Cavaillé, Jérôme (2008). "Non-coding RNAs in imprinted gene clusters". Biology of the Cell. 100 (3): 149–166. doi:10.1042/BC20070126. PMID 18271756.
- Huang, Hsien-Sung; Allen, John A.; Mabb, Angela M.; King, Ian F.; Miriyala, Jayalakshmi; Taylor-Blake, Bonnie; Sciaky, Noah; Dutton, J. Walter; et al. (2011). "Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons". Nature. 481 (7380): 185–189. doi:10.1038/nature10726. PMC 3257422. PMID 22190039.
- Horsthemke, Bernhard; Wagstaff, Joseph (2008). "Mechanisms of imprinting of the Prader-Willi/Angelman region". American Journal of Medical Genetics Part A. 146A (16): 2041–2052. doi:10.1002/ajmg.a.32364. PMID 18627066.
- Mabb, Angela M.; Judson, Matthew C.; Zylka, Mark J.; Philpot, Benjamin D. (2011). "Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes". Trends in Neurosciences. 34 (6): 293–303. doi:10.1016/j.tins.2011.04.001. PMC 3116240. PMID 21592595.
- Varon, Raymonda; Horn, Denise; Cohen, Monika Y.; Wagstaff, Joseph; Horsthemke, Bernhard; Buiting, Karin; Kroisel, Peter M.; Runte, Maren; Gillessen-Kaesbach, Gabriele (2004). "SNURF-SNRPN and UBE3A transcript levels in patients with Angelman syndrome". Human Genetics. 114 (6): 553–561. doi:10.1007/s00439-004-1104-z. PMID 15014980.
- Gray, T. A.; Saitoh, S; Nicholls, RD (1999). "An imprinted, mammalian bicistronic transcript encodes two independent proteins". Proceedings of the National Academy of Sciences. 96 (10): 5616–5621. doi:10.1073/pnas.96.10.5616. PMC 21909. PMID 10318933.
- Runte, Maren; Varon, Raymonda; Horn, Denise; Horsthemke, Bernhard; Buiting, Karin (2004). "Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader?Willi syndrome". Human Genetics. 116 (3): 228–230. doi:10.1007/s00439-004-1219-2. PMID 15565282.
- Kishore, S.; Khanna, A.; Zhang, Z.; Hui, J.; Balwierz, P. J.; Stefan, M.; Beach, C.; Nicholls, R. D.; et al. (2010). "The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing". Human Molecular Genetics. 19 (7): 1153–1164. doi:10.1093/hmg/ddp585. PMC 2838533. PMID 20053671.
- Wevrick, Rachel; Kerns, Julie A.; Francke, Uta (1994). "Identification of a novel paternally expressed gene in the Prader - Willi syndrome region". Human Molecular Genetics. 3 (10): 1877–1882. doi:10.1093/hmg/3.10.1877. PMID 7849716.
- Chamberlain, S. J.; Chen, P.-F.; Ng, K. Y.; Bourgois-Rocha, F.; Lemtiri-Chlieh, F.; Levine, E. S.; Lalande, M. (2010). "Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes". Proceedings of the National Academy of Sciences. 107 (41): 17668–73. doi:10.1073/pnas.1004487107. PMC 2955112. PMID 20876107.
- Reviewed in Dev Bio Le Meur, Elodie; Watrin, Françoise; Landers, Miguel; Sturny, Rachel; Lalande, Marc; Muscatelli, Françoise (2005). "Dynamic developmental regulation of the large non-coding RNA associated with the mouse 7C imprinted chromosomal region". Developmental Biology. 286 (2): 587–600. doi:10.1016/j.ydbio.2005.07.030. PMID 16126194.
- Chamberlain, S. J.; Lalande, M. (2010). "Angelman Syndrome, a Genomic Imprinting Disorder of the Brain". Journal of Neuroscience. 30 (30): 9958–9963. doi:10.1523/JNEUROSCI.1728-10.2010. PMC 6633366. PMID 20668179.
- Landers, M.; Calciano, MA; Colosi, D; Glatt-Deeley, H; Wagstaff, J; Lalande, M (2005). "Maternal disruption of Ube3a leads to increased expression of Ube3a-ATS in trans". Nucleic Acids Research. 33 (13): 3976–3984. doi:10.1093/nar/gki705. PMC 1178004. PMID 16027444.
- Perk, J.; Makedonski, K; Lande, L; Cedar, H; Razin, A; Shemer, R (2002). "The imprinting mechanism of the Prader-Willi/Angelman regional control center". The EMBO Journal. 21 (21): 5807–5814. doi:10.1093/emboj/cdf570. PMC 131067. PMID 12411498.
- Crampton, N.; Bonass, W. A.; Kirkham, J.; Rivetti, C.; Thomson, N. H. (2006). "Collision events between RNA polymerases in convergent transcription studied by atomic force microscopy". Nucleic Acids Research. 34 (19): 5416–5425. doi:10.1093/nar/gkl668. PMC 1636470. PMID 17012275.
- Callen, Benjamin P; Shearwin, Keith E; Egan, J.Barry (2004). "Transcriptional Interference between Convergent Promoters Caused by Elongation over the Promoter". Molecular Cell. 14 (5): 647–656. doi:10.1016/j.molcel.2004.05.010. PMID 15175159.
- Osato, N.; Suzuki, Y.; Ikeo, K.; Gojobori, T. (2006). "Transcriptional Interferences in cis Natural Antisense Transcripts of Humans and Mice". Genetics. 176 (2): 1299–1306. doi:10.1534/genetics.106.069484. PMC 1894591. PMID 17409075.
- Bird, Lynne M.; Tan, Wen-Hann; Bacino, Carlos A.; Peters, Sarika U.; Skinner, Steven A.; Anselm, Irina; Barbieri-Welge, Rene; Bauer-Carlin, Astrid; et al. (2011). "A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome". American Journal of Medical Genetics Part A. 155 (12): 2956–2963. doi:10.1002/ajmg.a.34297. PMC 3222728. PMID 22002941.