Catechol oxidase
Catechol oxidase is a copper oxidase that contains a type 3 di-copper cofactor and catalyzes the oxidation of ortho-diphenols into ortho-quinones coupled with the reduction of molecular oxygen to water. It is present in a variety of species of plants and fungi including Ipomoea batatas (sweet potato)[1] and Camellia sinensis (Indian tea leaf).[2] Metalloenzymes with type 3 copper centers are characterized by their ability to reversibly bind dioxygen at ambient conditions.[3] In plants, catechol oxidase plays a key role in enzymatic browning by catalyzing the oxidation of catechol to o-quinone in the presence of oxygen, which can rapidly polymerize to form the melanin that grants damaged fruits their dark brown coloration.
| Catechol oxidase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 1.10.3.1 | ||||||||
| CAS number | 9002-10-2 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| |||||||||
Biological Function

Polyphenol oxidases are a family of di-copper metalloenzymes that include tyrosinase and catechol oxidase.[4] In plants, both enzymes can catalyze the oxidation of ortho-diphenols substrates into their corresponding ortho-quinones. The key difference between the two related enzymes is that tyrosinase can catalyze the hydroxylation of monophenols to diphenols (monophenolase activity) as well as the oxidation of the o-diphenol to the o-quinone (diphenolase activity) whereas catechol oxidase only possesses diphenolase activity.[5]
When plant tissue is damaged, the chloroplast may rupture and release catechol oxidase into the plant cytoplasm, and vacuoles may also rupture, releasing stored catechol into the cytoplasm. The tissue damage also allows oxygen to penetrate into the cell. Thus, tissue damage facilitates the interaction of catechol oxidase with its substrate to produce o-benzoquinone, which can polymerize non-enzymatically to yield melanins that form an insoluble barrier for wound protection.[6]
Proteolytic Processing
Catechol oxidase is nuclear-encoded, and its N-terminal end contains a signal peptide that directs the protein to the chloroplast thylakoid lumen, where it can either be soluble or loosely associated with the thylakoid membrane.[7] Initially transcribed as a pro-enzyme, the catechol oxidase precursor undergoes two rounds of proteolytic processing and transport before it enters the thylakoid lumen.
Utilizing a [35S] methionine-labeled precursor protein, Sommer et al. elucidated a proteolytic processing pathway common to a variety of plants including pea (Pisum sativum), tomato (Lycopersicon esculentum), and maize (Zea mays).[8] The 67 kD precursor was imported into the stroma in an ATP-dependent manner where a stromal peptidase processes the precursor into a 62 kD intermediate. The translocation of this intermediate into the thylakoid lumen was light-dependent and results in the generation of the mature 59 kD enzyme.[9] Based on analysis of the precursor and mature catechol oxidase purified from Ipomoea batatas, proteolytic processing removes both the N-terminal transit peptide as well as a C-terminal domain that covers the enzyme active site.[10]
Enzyme Structure

The crystal structure of catechol oxidase purified from Ipomoea batatas has been resolved in its active form in both the oxidized Cu(II)-Cu(II) state and the reduced Cu(I)-Cu(I) state.[11] It is a globular, single domain monomeric enzyme that is approximately 55 by 45 by 45 Å in size and ellipsoid in shape. A four α-helix bundle comprises the enzyme core, which girds the active site containing the dicopper center.[12] The nitrogens on the imidazole side chains of His88, His109, and His118 coordinate with the first catalytic copper while the nitrogens on the imidazole side chains on His240, His244 and His274 coordinate with the second catalytic copper ion. In the oxidized Cu(II)-Cu(II) state, each copper ion possesses a four coordinate trigonal pyramidal geometry, with the three histidine residues and a bridging hydroxide molecule forming the four ligands on each copper ion. Comparing the reduced (Cu(I)-Cu(I)) state with the native (Cu(II)-Cu(II)) state of the enzyme, the key difference is the distance between the two copper centers. In the oxidized Cu(II)-Cu(II) state, the Cu-Cu distance is 3.3 Å while in the reduced Cu(I)-Cu(I) state, the distance increases to 4.4 Å.[1]
While the active site of both tyrosinase and catechol oxidase contain the di-copper center, variations in each enzyme’s respective structure result in differing activity. In catechol oxidase, a phenylalanine side-chain (Phe261) is above one of the copper centers and prevents the substrate from coordinating with both copper ions in the active site. This precludes the bidentate coordination complex necessary for di-phenolate hydroxylation characteristic of tyrosinase but absent in catechol oxidase.[13] Furthermore, His109 bound to one of the copper centers is also covalently linked with Cys192 through a thioether bridge.[14] This cysteine-histidine cross-linking may further restrain the enzyme active site from assuming the bidentate coordination complex readily formed in tyrosinase.
Catalytic Cycle and Mechanism
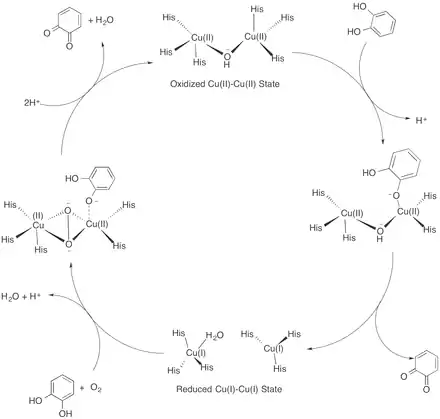
Although a crystal structure of catechol oxidase has been solved, questions concerning the exact mechanism of the reaction remain. One mechanism proposed by Eicken et al. is based on the crystal structure of catechol oxidase purified from Ipomoea batatas.[11] The catalytic cycle begins with the catechol oxidase in its native oxidized Cu(II)-Cu(II) state with a coordinated hydroxide ion bridging the two copper centers. As catechol enters the active site, a proton is abstracted from one of the alcohols. The catechol coordinates with a Cu(II) center in a monodentate fashion, displacing one of the coordinating histidine residues. The coordinated hydroxide ion abstracts another proton from catechol to form water, and the catechol is oxidized to o-quinone. The two resulting electrons reduce both copper centers to their Cu(I)-Cu(I) state. Dioxygen then binds one copper center, displacing the coordinated water molecule, and another molecule of catechol binds to the other copper center, displacing another histidine residue. This forms a complex in which one copper center has a tetragonal planar coordination with His240, His244 and the dioxygen molecule. The other copper center retains its initial tetragonal pyramidal geometry with dioxygen, His88 and His118 in the equatorial positions, and His109 in an axial position.[3] In this state, the enzyme active site is in a ternary catechol oxidase–O22−–catechol complex. Two electrons are transferred from the substrate to the dioxygen, followed by cleavage of the O–O bond. Water is released, and the second o-quinone product is formed together with the restoration of the initial Cu(II)-Cu(II) state to complete the catalytic cycle.[15]
This proposed catalytic cycle is supported by the experimental observation that stoichiometric amounts of o-quinone form after catechol addition to the enzyme, even when dioxygen is absent.[15] Furthermore, both the oxidized Cu(II)-Cu(II) state and the reduced Cu(I)-Cu(I) state were the two states identified by the crystal structure of Ipomoea batatas. The monodentate binding of catechol to the copper center was supported by the crystal structure of catechol oxidase bound with the bound-substrate analogue inhibitor phenylthiourea, which also binds to the copper center in a monodentate fashion.[11] However, one issue with this catalytic cycle is that the charge of the active site changes during the catalytic cycle from +1 to +3. This necessitates the presence of nearby bases that can store the protons; however, the X-ray crystal structure does not indicate the presence of any such bases as the histidine residues are coordinated with the copper centers.[16] Other catalytic cycles elucidated with DFT calculations and crystal structures have been proposed which maintain the same charge in the active site throughout the cycle and thus do not require nearby bases.[15][16] However, certain intermediates in the proposed cycle are not consistent with experimental findings such as that stoichiometric amounts of o-quinone can form after catechol addition in the absence of oxygen.[16]
Economic and Industrial Relevance
The oxidation of phenol substrates to their corresponding quinones are the primary cause of fruit and vegetable browning during ripening, handling, and processing.[17] Enzymatic browning affects the nutritional quality and appearance of fruits and produce. Over half of fruit losses are estimated to occur as a result of enzymatic browning, and tropical produce are particularly vulnerable to this reaction.[6] The loss of nutrients can occur due to the interaction of quinones, produced by the oxidation of diphenols, with the side chains of essential amino acids derived from plant proteins. In particular, thiol and amine functional groups on the side chains of amino acids are highly susceptible to quinone binding and alkylation.[18] The key role of catechol oxidase in enzymatic browning makes it a common target for inhibition. While a number of inhibitory strategies exist such as high temperature treatments(70-90 °C) to eliminate catechol oxidase catalytic activity,[6] a popular strategy is decreasing the pH with citric acid. Catechol oxidase is more catalytically active in the pH 4-8 range due to coordination of the histidine residues to the catalytic copper centers. The use of acids like citric acid to decrease the pH below this optimum range diminishes the binding of the enzyme to its active site copper because the protonation of histidine residues interferes with their ability to coordinate with the copper centers.[19]
Artificial Enzymes
New approaches to design artificial enzymes based on amino acids or peptides as characteristic molecular moieties have led to a significant expansion of the field of artificial enzymes or enzyme mimics. Recent results by the group of Rob Liskamp have shown that scaffolded histidine residues can be used as mimics of certain metalloproteins and -enzymes. The structural mimicry of certain copper proteins (e.g. hemocyanin, tyrosinase and catechol oxidase), containing type-3 copper binding sites, has been shown. This is a significant improvement since the use of scaffolded histidine residues is one step closer to the mimicry of enzymes by biologically relevant species.[20]
References
- Gerdemann C, Eicken C, Krebs B (March 2002). "The crystal structure of catechol oxidase: new insight into the function of type-3 copper proteins". Accounts of Chemical Research. 35 (3): 183–91. doi:10.1021/ar990019a. PMID 11900522.
- Halder J, Tamuli P, Bhaduri A (1998). "Isolation and characterization of polyphenol oxidase from Indian tea leaf (Camellia sinensis)". The Journal of Nutritional Biochemistry. 9 (2): 75–80. doi:10.1016/s0955-2863(97)00170-8.
- Koval IA, Gamez P, Belle C, Selmeczi K, Reedijk J (September 2006). "Synthetic models of the active site of catechol oxidase: mechanistic studies". Chemical Society Reviews. 35 (9): 814–40. doi:10.1039/b516250p. PMID 16936929.
- Dey SK, Mukherjee A (2016). "Catechol oxidase and phenoxazinone synthase: Biomimetic functional models and mechanistic studies". Coordination Chemistry Reviews. 310: 80–115. doi:10.1016/j.ccr.2015.11.002.
- Gerdemann C, Eicken C, Magrini A, Meyer HE, Rompel A, Spener F, Krebs B (July 2001). "Isozymes of Ipomoea batatas catechol oxidase differ in catalase-like activity". Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 1548 (1): 94–105. doi:10.1016/s0167-4838(01)00219-9. PMID 11451442.
- Queiroz C, Lopes ML, Fialho E, Valente-Mesquita VL (2008). "Polyphenol Oxidase: Characteristics and Mechanisms of Browning Control". Food Reviews International. 24 (4): 361–375. doi:10.1080/87559120802089332.
- Marusek CM, Trobaugh NM, Flurkey WH, Inlow JK (January 2006). "Comparative analysis of polyphenol oxidase from plant and fungal species". Journal of Inorganic Biochemistry. 100 (1): 108–23. doi:10.1016/j.jinorgbio.2005.10.008. PMID 16332393.
- Sommer A, Ne'eman E, Steffens JC, Mayer AM, Harel E (August 1994). "Import, targeting, and processing of a plant polyphenol oxidase". Plant Physiology. 105 (4): 1301–11. doi:10.1104/pp.105.4.1301. PMC 159463. PMID 7972497.
- Mayer AM (November 2006). "Polyphenol oxidases in plants and fungi: going places? A review". Phytochemistry. 67 (21): 2318–31. doi:10.1016/j.phytochem.2006.08.006. PMID 16973188.
- Flurkey WH, Inlow JK (December 2008). "Proteolytic processing of polyphenol oxidase from plants and fungi". Journal of Inorganic Biochemistry. 102 (12): 2160–70. doi:10.1016/j.jinorgbio.2008.08.007. PMID 18829115.
- Eicken C, Krebs B, Sacchettini JC (December 1999). "Catechol oxidase - structure and activity". Current Opinion in Structural Biology. 9 (6): 677–83. doi:10.1016/s0959-440x(99)00029-9. PMID 10607672.
- Klabunde T, Eicken C, Sacchettini JC, Krebs B (December 1998). "Crystal structure of a plant catechol oxidase containing a dicopper center". Nature Structural Biology. 5 (12): 1084–90. doi:10.1038/4193. PMID 9846879.
- Lucas HR, Karlin KD (2009). "Chapter 9: Copper-Carbon Bonds in Mechanistic and Structural Probing of Proteins as well as in Situations where Copper is a Catalytic or Receptor Site". In Sigel A, Sigel H, Sigel RK (eds.). Metal-carbon bonds in enzymes and cofactors. Cambridge, UK: RSC Publishing. p. 304. ISBN 978-1-84755-915-9.
- Virador VM, Reyes Grajeda JP, Blanco-Labra A, Mendiola-Olaya E, Smith GM, Moreno A, Whitaker JR (January 2010). "Cloning, sequencing, purification, and crystal structure of Grenache (Vitis vinifera) polyphenol oxidase". Journal of Agricultural and Food Chemistry. 58 (2): 1189–201. doi:10.1021/jf902939q. PMID 20039636.
- Siegbahn PE (July 2004). "The catalytic cycle of catechol oxidase". Journal of Biological Inorganic Chemistry. 9 (5): 577–90. doi:10.1007/s00775-004-0551-2. PMID 15185133.
- Güell M, Siegbahn PE (November 2007). "Theoretical study of the catalytic mechanism of catechol oxidase". Journal of Biological Inorganic Chemistry. 12 (8): 1251–64. doi:10.1007/s00775-007-0293-z. PMID 17891425.
- Martinez M, Whitaker JR (June 1995). "The biochemistry and control of enzymatic browning". Trends in Food Science & Technology. 6 (6): 195–200. doi:10.1016/S0924-2244(00)89054-8.
- Mathesis G, Whitaker JR (September 1984). "Modification of Proteins by Polyphenol Oxidase and Peroxidase and their Products". Journal of Food Biochemistry. 8 (3): 137–162. doi:10.1111/j.1745-4514.1984.tb00322.x.
- Yoruk R, Marshall MR (November 2003). "Physicochemical Properties and Function of Plant Polyphenol Oxidase: A Review". Journal of Food Biochemistry. 27 (5): 361–422. doi:10.1111/j.1745-4514.2003.tb00289.x.
- Albada HB, Soulimani F, Weckhuysen BM, Liskamp RM (December 2007). "Scaffolded amino acids as a close structural mimic of type-3 copper binding sites". Chemical Communications (Cambridge, England) (46): 4895–7. doi:10.1039/B709400K. PMID 18361361.
External links
- Catechol+Oxidase at the US National Library of Medicine Medical Subject Headings (MeSH)