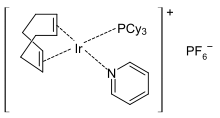
Crabtree's catalyst
Crabtree's catalyst is an organoiridium compound with the formula [C8H12IrP(C6H11)3C5H5N]PF6. It is a homogeneous catalyst for hydrogenation and hydrogen-transfer reactions, developed by Robert H. Crabtree. This air stable orange solid is commercially available and known for its directed hydrogenation to give trans stereoselectivity with respective of directing group.[2][3]
 | |
 | |
| Names | |
|---|---|
| IUPAC name
(SP-4)tris(cyclohexyl)phosphane | |
| Identifiers | |
3D model (JSmol) |
|
| ChemSpider | |
| ECHA InfoCard | 100.164.161 |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C31H50F6IrNP2 | |
| Molar mass | 804.9026 g/mol |
| Appearance | Yellow microcrystals |
| Melting point | 150 °C (302 °F; 423 K) (decomposes)[1] |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
Structure and synthesis
The complex has a square planar molecular geometry, as expected for a d8 complex. It is prepared from cyclooctadiene iridium chloride dimer.[4]
Reactivity
Crabtree’s catalyst is effective for the hydrogenations of mono-, di-, tri-, and tetra-substituted substrates. Whereas Wilkinson’s catalyst and the Schrock–Osborn catalyst do not catalyze the hydrogenation of a tetrasubstituted olefin, Crabtree’s catalyst does so to at high turnover frequencies (table).[2][5]
Turnover frequencies Substrate Wilkinson's catalyst Schrock–Osborn catalyst Crabtree's catalyst 1-Hexene 650 4000 6400 Cyclohexene 700 10 4500 1-Methylcyclohexene 13 — 3800 2,3-Dimethyl-2-butene — — 4000
The catalyst is reactive at room temperature.[1] The reaction is robust without drying solvents or meticulous deoxygenation of the hydrogen. The catalyst is tolerant of weakly basic functional groups such as ester, but not alcohols (see below) or amines.[2] The catalyst is sensitive to proton-bearing impurities.[6]
The catalyst becomes irreversibly deactivated after about ten minutes at room temperature, signaled by appearance of yellow color. One deactivation process involves formation of hydride-bridged dimers.[7] As a consequence, Crabtree's Catalyst is usually used in very low catalyst loading.
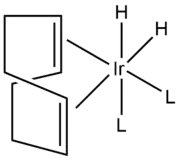
Other catalytic functions: isotope exchange and isomerization
Besides hydrogenation, the catalyst catalyzes the isomerization and hydroboration of alkenes.[1]
Crabtree's catalyst is used in isotope exchange reactions. More specifically, it catalyzes the direct exchange of a hydrogen atom with its isotopes deuterium and tritium, without the use of an intermediate.[8] It has been shown that isotope exchange with Crabtree’s catalyst is highly regioselective.[9][10]
Influence of directing functional groups
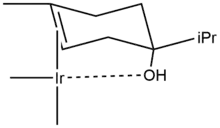
The hydrogenation of a terpen-4-ol demonstrates the ability of compounds with directing groups (the –OH group) to undergo diastereoselective hydrogenation. With palladium on carbon in ethanol the product distribution is 20:80 favoring the cis isomer (2B in Scheme 1). The polar side (with the hydroxyl group) interacts with the solvent. This is due to slight haptophilicity, an effect in which a functional group binds to the surface of a heterogeneous catalyst and directs the reaction.[11][12] In cyclohexane as solvent, the distribution changes to 53:47 because haptophilicity is no long present (there is no directing group on cyclohexane). The distribution changes completely in favor of the cis isomer 2A when Crabtree's catalyst is used in dichloromethane. This selectivity is both predictable and practically useful.[13] Carbonyl groups are also known to direct the hydrogenation by the Crabtree catalyst to be highly regioselective.[14][15][16]

The directing effect that causes the stereoselectivity of hydrogenation of terpen-4-ol with Crabtree’s catalyst is shown below.
History
Crabtree and graduate student George Morris discovered this catalyst in the 1970s while working on iridium analogues of Wilkinson's rhodium-based catalyst at the Institut de Chimie des Substances Naturelles at Gif-sur-Yvette, near Paris.
Previous hydrogenation catalysts included Wilkinson’s catalyst and a cationic rhodium(I) complex with two phosphine groups developed by Osborn and Schrock.[17] These catalysts accomplished hydrogenation through displacement; after hydrogen addition across the metal, a solvent or a phosphine group dissociated from the rhodium metal so the olefin to be hydrogenated could gain access to the active site.[2] This displacement occurs quickly for rhodium complexes but occurs barely at all for iridium complexes.[18] Because of this, research at the time focused on rhodium compounds instead of compounds involving transition metals of the third row, like iridium. Wilkinson, Osborn, and Schrock also only used coordinating solvents.[19]
Crabtree noted that the ligand dissociation step does not occur in heterogeneous catalysis, and so posited that this step was limiting in homogeneous systems.[2] They sought catalysts with “irreversibly creat[ed] active sites in a noncoordinating solvent.” This led to the development of the Crabtree catalyst, and use of the solvent CH2Cl2.
References
- Crabtree, R. H. (2001). "(1,5-Cyclooctadiene)(tricyclohexylphosphine)(pyridine)iridium(I) Hexafluorophosphate". e-EROS Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rc290m.pub4.
- Crabtree, R. H. (1979). "Iridium compounds in catalysis". Acc. Chem. Res. 12 (9): 331–337. doi:10.1021/ar50141a005.
- Brown, J. M. (1987). "Directed Homogeneous Hydrogenation". Angew. Chem. Int. Ed. 26 (3): 190–203. doi:10.1002/anie.198701901.
- Crabtree, R. H.; Morris, G. E. (1977). "Some diolefin complexes of Iridium(I) and a trans-Influence Series for the Complexes [IrCl(cod)L]". J. Organomet. Chem. 135 (3): 395–403. doi:10.1016/S0022-328X(00)88091-2.
- White, M. (2002-10-15). "Hydrogenation" (PDF). Retrieved 2014-12-01.
- Xu, Yingjian; Mingos, D. Michael P.; Brown, John M. (2008). "Crabtree's catalyst revisited; Ligand effects on stability and durability". Chem. Comm. 2008 (2): 199–201. doi:10.1039/b711979h. PMID 18092086.
- Crabtree, R.; Felkin, H.; Morris, G. (1977). "Cationic iridium diolefin complexes as alkene hydrogenation catalysts and the isolation of some related hydrido complexes". J. Organomet. Chem. 141: 205–215. doi:10.1016/S0022-328X(00)92273-3.
- Schou, S. (2009). "The effect of adding Crabtree's catalyst to rhodium black in direct hydrogen isotope exchange reactions". Journal of Labelled Compounds and Radiopharmaceuticals. 52: 376–381. doi:10.1002/jlcr.1612.
- Valsborg, J.; Sorensen, L.; Foged, C. (2001). "Organoiridium catalysed hydrogen isotope exchange of benzamide derivatives". Journal of Labelled Compounds and Radiopharmaceuticals. 44: 209–214. doi:10.1002/jlcr.446.
- Hesk, D.; Das, P.; Evans, B. (1995). "Deuteration of acetanilides and other substituted aromatics using [Ir(COD)(Cy3P)(Py)]PF6 as catalyst". Journal of Labelled Compounds and Radiopharmaceuticals. 36 (5): 497–502. doi:10.1002/jlcr.2580360514.
- Thompson, H.; Naipawer, R. (1973). "Stereochemical control of reductions. III. Approach to group haptophilicities". J. Am. Chem. Soc. 95 (19): 6379–6386. doi:10.1021/ja00800a036.
- Rowlands, G. (2002-01-01). "Hydrogenation" (PDF). Retrieved 2014-12-01.
- Brown, J. (1987). "Directed Homogeneous Hydrogenation [New Synthetic Methods (65)]". Angew. Chem. Int. Ed. Engl. 26 (3): 190–203. doi:10.1002/anie.198701901.
- Schultz, A.; McCloskey, P. (1985). "Carboxamide and carbalkoxy group directed stereoselective iridium-catalyzed homogeneous olefin hydrogenations". J. Org. Chem. 50 (26): 5905–5907. doi:10.1021/jo00350a105.
- Crabtree, R. H.; Davis, M. W. (1986). "Directing effects in homogeneous hydrogenation with [Ir(cod)(PCy3)(py)]PF6". J. Org. Chem. 51 (14): 2655–2661. doi:10.1021/jo00364a007.
- Crabtree, R.; Davis, M. (1983). "Occurrence and origin of a pronounced directing effect of a hydroxyl group in hydrogenation with [Ir(cod)P(C6H11)3(py)]PF6". Organometallics. 2: 681–682. doi:10.1021/om00077a019.
- Schrock, R.; Osborn, J. A. (1976). "Catalytic hydrogenation using cationic rhodium complexes. I. Evolution of the catalytic system and the hydrogenation of olefins". J. Am. Chem. Soc. 98 (8): 2134–2143. doi:10.1021/ja00424a020.
- Osborn, J.; Shapley, J. (1970). "Rapid intramolecular rearrangements in pentacoordinate transition metal compounds. Rearrangement mechanism of some fluxional iridium(I) complexes". J. Am. Chem. Soc. 92 (23): 6976–6978. doi:10.1021/ja00726a047.
- Young, J.; Wilkinson, G. (1966). "The preparation and properties of tris(triphenylphosphine)halogenorhodium(I) and some reactions thereof including catalytic homogeneous hydrogenation of olefins and acetylenes and their derivatives". J. Chem. Soc. A. 1966: 1711. doi:10.1039/J19660001711.