DNA demethylation
In mammals, DNA demethylation causes replacement of 5-methylcytosine (5mC) in a DNA sequence by cytosine (C) (see figure of 5mC and C). DNA demethylation can occur by an active process at the site of a 5mC in a DNA sequence or, in replicating cells, by preventing addition of methyl groups to DNA so that the replicated DNA will largely have cytosine in the DNA sequence (5mC will be diluted out).

Methylated cytosine is frequently present in the linear DNA sequence where a cytosine is followed by a guanine in a 5' → 3' direction (a CpG site). In mammals, DNA methyltransferases (which add methyl groups to DNA bases) exhibit a strong sequence preference for cytosines at (CpG sites).[1] There appear to be more than 20 million CpG dinucleotides in the human genome (see genomic distribution). In mammals, on average, 70% to 80% of CpG cytosines are methylated,[2] though the level of methylation varies with different tissues. Methylated cytosines often occur in groups or CpG islands within the promoter regions of genes, where such methylation may reduce or silence gene expression (see gene expression). Methylated cytosines in the gene body, however, are positively correlated with expression.[3]
Almost 100% DNA demethylation occurs by a combination of passive dilution and active enzymatic removal during the reprogramming that occurs in early embryogenesis and in gametogenesis. Another large demethylation, of about 3% of all genes, can occur by active demethylation in neurons during formation of a strong memory.[4] After surgery, demethylations are found in peripheral blood mononuclear cells at sites annotated to immune system genes.[5] Demethylations also occur during the formation of cancers.[6] During global DNA hypomethylation of tumor genomes, there is a minor to moderate reduction of the number of methylated cytosines (5mC) amounting to a loss of about 5% to 20% on average of the 5mC bases.[7]
Embryonic development
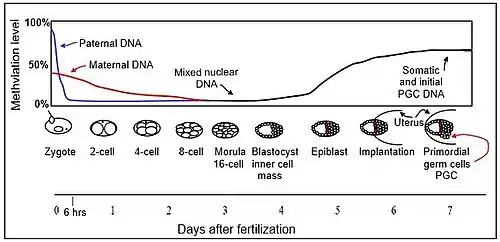
Early embryonic development
The mouse sperm genome is 80–90% methylated at its CpG sites in DNA, amounting to about 20 million methylated sites. After fertilization, the paternal chromosome is almost completely demethylated in six hours by an active process, before DNA replication (blue line in Figure).
Demethylation of the maternal genome occurs by a different process. In the mature oocyte, about 40% of its CpG sites in DNA are methylated. While somatic cells of mammals have three main DNA methyltransferases (which add methyl groups to cytosines at CpG sites), DNMT1, DNMT3A, and DNMT3B, in the pre-implantation embryo up to the blastocyst stage (see Figure), the only methyltransferase present is an isoform of DNMT1 designated DNMT1o.[8] DNMT1o has an alternative oocyte-specific promoter and first exon (exon 1o) located 5' of the somatic and spermatocyte promoters. As reviewed by Howell et al.,[8] DNMT1o is sequestered in the cytoplasm of mature oocytes and in 2-cell and 4-cell embryos, but at the 8-cell stage is only present in the nucleus. At the 16 cell stage (the morula) DNMT1o is again found only in the cytoplasm. It appears that demethylation of the maternal chromosomes largely takes place by blockage of the methylating enzyme DNMT1o from entering the nucleus except briefly at the 8 cell stage. The maternal-origin DNA thus undergoes passive demethylation by dilution of the methylated maternal DNA during replication (red line in Figure). The morula (at the 16 cell stage), has only a small amount of DNA methylation (black line in Figure).
DNMT3b begins to be expressed in the blastocyst.[9] Methylation begins to increase at 3.5 days after fertilization in the blastocyst, and a large wave of methylation then occurs on days 4.5 to 5.5 in the epiblast, going from 12% to 62% methylation, and reaching maximum level after implantation in the uterus.[10] By day seven after fertilization, the newly formed primordial germ cells (PGC) in the implanted embryo segregate from the remaining somatic cells. At this point the PGCs have about the same level of methylation as the somatic cells.
Gametogenesis
The newly formed primordial germ cells (PGC) in the implanted embryo devolve from the somatic cells. At this point the PGCs have high levels of methylation. These cells migrate from the epiblast toward the gonadal ridge. As reviewed by Messerschmidt et al.,[11] the majority of PGCs are arrested in the G2 phase of the cell cycle, while they migrate toward the hindgut during embryo days 7.5 to 8.5. Then demethylation of the PGCs takes place in two waves.[11] At day 9.5 the primordial germ cells begin to rapidly replicate going from about 200 PGCs at embryo day 9.5 to about 10,000 PGCs at day 12.5.[12] During days 9.5 to 12.5 DNMT3a and DNMT3b are repressed and DNMT1 is present in the nucleus at a high level. But DNMT1 is unable to methylate cytosines during days 9.5 to 12.5 because the UHRF1 gene (also known as NP95) is repressed and UHRF1 is an essential protein needed to recruit DNMT1 to replication foci where maintenance DNA methylation takes place.[12] This is a passive, dilution form of demethylation.
In addition, from embryo day 9.5 to 13.5 there is an active form of demethylation. As indicated below in "Molecular stages of active reprogramming," two enzymes are central to active demethylation. These are a ten-eleven translocation methylcytosine dioxygenase (TET) and thymine-DNA glycosylase (TDG). One particular TET enzyme, TET1, and TDG are present at high levels from embryo day 9.5 to 13.5,[12] and are employed in active demethylation during gametogenesis.[11] PGC genomes display the lowest levels of DNA methylation of any cells in the entire life cycle of the mouse at embryonic day 13.5. [13]
Learning and Memory
Learning and memory have levels of permanence, differing from other mental processes such as thought, language, and consciousness, which are temporary in nature. Learning and memory can be either accumulated slowly (multiplication tables) or rapidly (touching a hot stove), but once attained, can be recalled into conscious use for a long time. Rats subjected to one instance of contextual fear conditioning create an especially strong long-term memory. At 24 hours after training, 9.17% of the genes in the genomes of rat hippocampus neurons were found to be differentially methylated. This included more than 2,000 differentially methylated genes at 24 hours after training, with over 500 genes being demethylated.[4] Similar results to that in the rat hippocampus were also obtained in mice with contextual fear conditioning.[14]
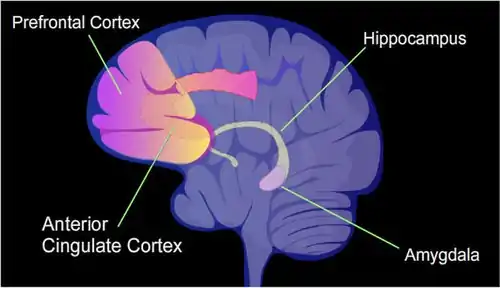
The hippocampus region of the brain is where contextual fear memories are first stored (see figure of the brain, this section), but this storage is transient and does not remain in the hippocampus. In rats contextual fear conditioning is abolished when the hippocampus is subjected to hippocampectomy just one day after conditioning, but rats retain a considerable amount of contextual fear when hippocampectomy is delayed by four weeks.[15] In mice, examined at 4 weeks after conditioning, the hippocampus methylations and demethylations were reversed (the hippocampus is needed to form memories but memories are not stored there) while substantial differential CpG methylation and demethylation occurred in cortical neurons during memory maintenance. There were 1,223 differentially methylated genes in the anterior cingulate cortex of mice four weeks after contextual fear conditioning. Thus, while there were many methylations in the hippocampus shortly after memory was formed, all these hippocampus methylations were demethylated as soon as four weeks later.
Demethylation in Cancer
The human genome contains about 28 million CpG sites, and roughly 60% of the CpG sites are methylated at the 5 position of the cytosine. [16] During formation of a cancer there is an average reduction of the number of methylated cytosines of about 5% to 20%,[17] or about 840,00 to 3.4 million demethylations of CpG sites.
DNMT1 methylates CpGs on hemi-methylated DNA during DNA replication. Thus, when a DNA strand has a methylated CpG, and the newly replicated strand during semi-conservative replication lacks a methyl group on the complementary CpG, DNMT1 is normally recruited to the hemimethylated site and adds a methyl group to cytosine in the newly synthesized CpG. However, recruitment of DNMT1 to hemimethylated CpG sites during DNA replication depends on the UHRF1 protein. If UHRF1 does not bind to a hemimethylated CpG site, then DNMT1 is not recruited and cannot methylate the newly synthesized CpG site. The arginine methyltransferase PRMT6 regulates DNA methylation by methylating the arginine at position 2 of histone 3 (H3R2me2a).[18] (See Protein methylation#Arginine.) In the presence of H3R2me2a UHRF1 can not bind to a hemimethylated CpG site, and then DNMT1 is not recruited to the site, and the site remains hemimethylated. Upon further rounds of replication the methylated CpG is passively diluted out. PRMT6 is frequently overexpressed in many types of cancer cells.[19] The overexpression of PRMT6 may be a source of DNA demethylation in cancer.
Molecular stages of active reprogramming
Three molecular stages are required for actively, enzymatically reprogramming the DNA methylome. Stage 1: Recruitment. The enzymes needed for reprogramming are recruited to genome sites that require demethylation or methylation. Stage 2: Implementation. The initial enzymatic reactions take place. In the case of methylation, this is a short step that results in the methylation of cytosine to 5-methylcytosine. Stage 3: Base excision DNA repair. The intermediate products of demethylation are catalysed by specific enzymes of the base excision DNA repair pathway that finally restore cystosine in the DNA sequence.
Stage 2 of active demethylation
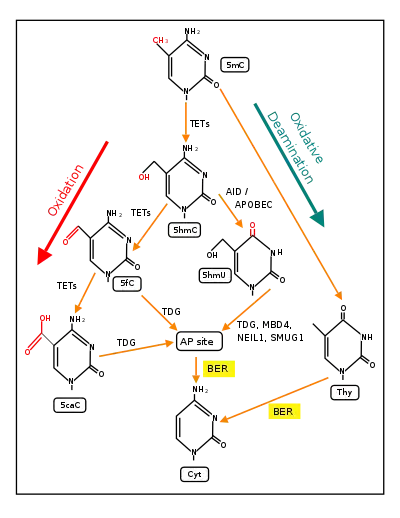
Demethylation of 5-methylcytosine to generate 5-hydroxymethylcytosine (5hmC) very often initially involves oxidation of 5mC (see Figure in this section) by ten-eleven translocation methylcytosine dioxygenases (TET enzymes).[21] The molecular steps of this initial demethylation are shown in detail in TET enzymes. In successive steps (see Figure) TET enzymes further hydroxylate 5hmC to generate 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Thymine-DNA glycosylase (TDG) recognizes the intermediate bases 5fC and 5caC and excises the glycosidic bond resulting in an apyrimidinic site (AP site). This is followed by base excision repair (stage 3). In an alternative oxidative deamination pathway, 5hmC can be oxidatively deaminated by APOBEC (AID/APOBEC) deaminases to form 5-hydroxymethyluracil (5hmU). Also, 5mC can be converted to thymine (Thy). 5hmU can be cleaved by TDG, MBD4, NEIL1 or SMUG1. AP sites and T:G mismatches are then repaired by base excision repair (BER) enzymes to yield cytosine (Cyt). The TET family of dioxygenases are employed in the most frequent type of demethylation reactions.[21]
TET family
TET dioxygenase isoforms include at least two isoforms of TET1, one of TET2 and three isoforms of TET3.[22][23] The full-length canonical TET1 isoform appears virtually restricted to early embryos, embryonic stem cells and primordial germ cells (PGCs). The dominant TET1 isoform in most somatic tissues, at least in the mouse, arises from alternative promoter usage which gives rise to a short transcript and a truncated protein designated TET1s. The isoforms of TET3 are the full length form TET3FL, a short form splice variant TET3s, and a form that occurs in oocytes and neurons designated TET3o. TET3o is created by alternative promoter use and contains an additional first N-terminal exon coding for 11 amino acids. TET3o only occurs in oocytes and neurons and is not expressed in embryonic stem cells or in any other cell type or adult mouse tissue tested. Whereas TET1 expression can barely be detected in oocytes and zygotes, and TET2 is only moderately expressed, the TET3 variant TET3o shows extremely high levels of expression in oocytes and zygotes, but is nearly absent at the 2-cell stage. It is possible that TET3o, high in neurons, oocytes and zygotes at the one cell stage, is the major TET enzyme utilized when very large scale rapid demethylations occur in these cells.
Stage 1 of demethylation - recruitment of TET to DNA
The TET enzymes do not specifically bind to 5-methylcytosine except when recruited. Without recruitment or targeting, TET1 predominantly binds to high CG promoters and CpG islands (CGIs) genome-wide by its CXXC domain that can recognize un-methylated CGIs.[24] TET2 does not have an affinity for 5-methylcytosine in DNA.[25] The CXXC domain of the full-length TET3, which is the predominant form expressed in neurons, binds most strongly to CpGs where the C was converted to 5-carboxycytosine (5caC). However, it also binds to un-methylated CpGs.[23]
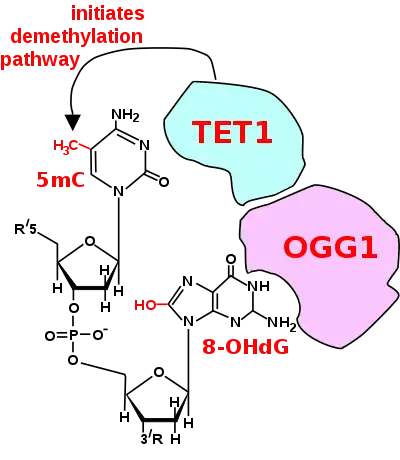
For a TET enzyme to initiate demethylation it must first be recruited to a methylated CpG site in DNA. Two of the proteins shown to recruit a TET enzyme to a methylated cytosine in DNA are OGG1 (see figure Initiation of DNA demethylation at a CpG site)[26] and EGR1.[27]
OGG1
Oxoguanine glycosylase (OGG1) catalyses the first step in base excision repair of the oxidatively damaged base 8-OHdG. OGG1 finds 8-OHdG by sliding along the linear DNA at 1,000 base pairs of DNA in 0.1 seconds.[28] OGG1 very rapidly finds 8-OHdG. OGG1 proteins bind to oxidatively damaged DNA with a half maximum time of about 6 seconds.[29] When OGG1 finds 8-OHdG it changes conformation and complexes with 8-OHdG in its binding pocket.[30] OGG1 does not immediately act to remove the 8-OHdG. Half maximum removal of 8-OHdG takes about 30 minutes in HeLa cells in vitro,[31] or about 11 minutes in the livers of irradiated mice.[32] DNA oxidation by reactive oxygen species preferentially occurs at a guanine in a methylated CpG site, because of a lowered ionization potential of guanine bases adjacent to 5-methylcytosine.[33] TET1 binds (is recruited to) the OGG1 bound to 8-OHdG (see figure).[26] This likely allows TET1 to demethylate an adjacent methylated cytosine. When human mammary epithelial cells (MCF-10A) were treated with H2O2, 8-OHdG increased in DNA by 3.5-fold and this caused about 80% demethylation of the 5-methylcytosines in the MCF-10A genome.[26]
EGR1
The gene early growth response protein 1 (EGR1) is an immediate early gene (IEG). EGR1 can rapidly be induced by neuronal activity.[34] The defining characteristic of IEGs is the rapid and transient up-regulation—within minutes—of their mRNA levels independent of protein synthesis.[35] In adulthood, EGR1 is expressed widely throughout the brain, maintaining baseline expression levels in several key areas of the brain including the medial prefrontal cortex, striatum, hippocampus and amygdala.[35] This expression is linked to control of cognition, emotional response, social behavior and sensitivity to reward.[35] EGR1 binds to DNA at sites with the motifs 5′-GCGTGGGCG-3′ and 5'-GCGGGGGCGG-3′ and these motifs occur primarily in promoter regions of genes.[34] The short isoform TET1s is expressed in the brain. EGR1 and TET1s form a complex mediated by the C-terminal regions of both proteins, independently of association with DNA.[34] EGR1 recruits TET1s to genomic regions flanking EGR1 binding sites.[34] In the presence of EGR1, TET1s is capable of locus-specific demethylation and activation of the expression of downstream genes regulated by EGR1.[34]
DNA demethylation intermediate 5hmC
As indicated in the Figure above, captioned "Demethylation of 5-methylcytosine," the first step in active demethylation is a TET oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). The demethylation process, in some tissues and at some genome locations, may stop at that point. As reviewed by Uribe-Lewis et al.,[36] in addition to being an intermediate in active DNA demethylation, 5hmC is often a stable DNA modification. Within the genome, 5hmC is located at transcriptionally active genes, regulatory elements and chromatin associated complexes. In particular, 5hmC is dynamically changed and positively correlated with active gene transcription during cell lineage specification, and high levels of 5hmC are found in embryonic stem cells and in the central nervous system.[37] In humans, defective 5-hydroxymethylating activity is associated with a phenotype of lymphoproliferation, immunodeficiency and autoimmunity.[38]
Stage 3 base excision repair
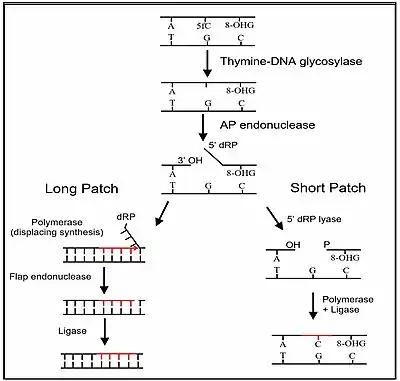
The third stage of DNA demethylation is removal of the intermediate products of demethylation generated by a TET enzyme by base excision repair. As indicated above in Stage 2, after 5mC is first oxidized by a TET to form 5hmC, further oxidation of 5hmC by TET yields 5fC and oxidation of 5fC by TET yields 5caC. Both 5fC and 5caC are recognized by a DNA glycosylase, TDG, a base excision repair enzyme, as an abnormal base. As shown in the Figure in this section, TDG removes the abnormal base (e.g. 5fC) while leaving the sugar-phosphate backbone intact, creating an apurinic/apyrimidinic site, commonly referred to as an AP site. In this Figure, the 8-OHdG is left in the DNA, since it may have been present when OGG1 attracted TET1 to the CpG site with a methylated cytosine. After an AP site is formed, AP endonuclease creates a nick in the phosphodiester backbone of the AP site that was formed when the TDG DNA glycosylase removed the 5fC or 5caC. The human AP endonuclease incises DNA 5′ to the AP site by a hydrolytic mechanism, leaving a 3′-hydroxyl and a 5′-deoxyribose phosphate (5' dRP) residue.[39] This is followed by either short patch or long patch repair. In short patch repair, 5′ dRP lyase trims the 5′ dRP end to form a phosphorylated 5′ end. This is followed by DNA polymerase β (pol β) adding a single cytosine to pair with the pre-existing guanine in the complementary strand and then DNA ligase to seal the cut strand. In long patch repair, DNA synthesis is thought to be mediated by polymerase δ and polymerase ε performing displacement synthesis to form a flap. Pol β can also perform long-patch displacement synthesis. Long-patch synthesis typically inserts 2–10 new nucleotides. Then flap endonuclease removes the flap, and this is followed by DNA ligase to seal the strand. At this point there has been a complete replacement of the 5-methylcytosine by cytosine (demethylation) in the DNA sequence.
Demethylation after exercise
Physical exercise has well established beneficial effects on learning and memory (see Neurobiological effects of physical exercise). BDNF is a particularly important regulator of learning and memory.[40] As reviewed by Fernandes et al.,[41] in rats, exercise enhances the hippocampus expression of the gene Bdnf, which has an essential role in memory formation. Enhanced expression of Bdnf occurs through demethylation of its CpG island promoter at exon IV[41] and this demethylation depends on steps illustrated in the two figures.[20]
Demethylation after exposure to traffic related air pollution
In a panel of healthy adults, negative associations were found between total DNA methylation and exposure to traffic related air pollution. DNA methylation levels were associated both with recent and chronic exposure to Black Carbon as well as benzene. [42]
Peripheral sensory neuron regeneration
After injury, neurons in the adult peripheral nervous system can switch from a dormant state with little axonal growth to robust axon regeneration. DNA demethylation in mature mammalian neurons removes barriers to axonal regeneration.[43] This demethylation, in regenerating mouse peripheral neurons, depends upon TET3 to generate 5-hydroxymethylcytosine (5hmC) in DNA.[43][44] 5hmC was altered in a large set of regeneration-associated genes (RAGs), including well-known RAGs such as Atf3, Bdnf, and Smad1, that regulate the axon growth potential of neurons.[44]
References
- Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, Gnirke A, Meissner A (December 2011). "Genomic distribution and inter-sample variation of non-CpG methylation across human cell types". PLOS Genet. 7 (12): e1002389. doi:10.1371/journal.pgen.1002389. PMC 3234221. PMID 22174693.
- Jabbari K, Bernardi G (May 2004). "Cytosine methylation and CpG, TpG (CpA) and TpA frequencies". Gene. 333: 143–9. doi:10.1016/j.gene.2004.02.043. PMID 15177689.
- Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G (October 2014). "Gene body methylation can alter gene expression and is a therapeutic target in cancer". Cancer Cell. 26 (4): 577–90. doi:10.1016/j.ccr.2014.07.028. PMC 4224113. PMID 25263941.
- Duke CG, Kennedy AJ, Gavin CF, Day JJ, Sweatt JD (July 2017). "Experience-dependent epigenomic reorganization in the hippocampus". Learn. Mem. 24 (7): 278–288. doi:10.1101/lm.045112.117. PMC 5473107. PMID 28620075.
- Sadahiro R, Knight B, James F, Hannon E, Charity J, Daniels IR, Burrage J, Knox O, Crawford B, Smart NJ, Mill J (April 2020). "Major surgery induces acute changes in measured DNA methylation associated with immune response pathways". Sci Rep. 10 (1): 5743. doi:10.1038/s41598-020-62262-x. PMC 7113299. PMID 32238836.
- Ehrlich M (December 2009). "DNA hypomethylation in cancer cells". Epigenomics. 1 (2): 239–59. doi:10.2217/epi.09.33. PMC 2873040. PMID 20495664.
- Pfeifer GP (April 2018). "Defining Driver DNA Methylation Changes in Human Cancer". Int J Mol Sci. 19 (4): 1166. doi:10.3390/ijms19041166. PMC 5979276. PMID 29649096.
- Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR (March 2001). "Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene". Cell. 104 (6): 829–38. doi:10.1016/s0092-8674(01)00280-x. PMID 11290321.
- Watanabe D, Suetake I, Tada T, Tajima S (October 2002). "Stage- and cell-specific expression of Dnmt3a and Dnmt3b during embryogenesis". Mech. Dev. 118 (1–2): 187–90. doi:10.1016/s0925-4773(02)00242-3. PMID 12351185.
- Auclair G, Guibert S, Bender A, Weber M (2014). "Ontogeny of CpG island methylation and specificity of DNMT3 methyltransferases during embryonic development in the mouse". Genome Biol. 15 (12): 545. doi:10.1186/s13059-014-0545-5. PMC 4295324. PMID 25476147.
- Messerschmidt DM, Knowles BB, Solter D (April 2014). "DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos". Genes Dev. 28 (8): 812–28. doi:10.1101/gad.234294.113. PMC 4003274. PMID 24736841.
- Kagiwada S, Kurimoto K, Hirota T, Yamaji M, Saitou M (February 2013). "Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice". EMBO J. 32 (3): 340–53. doi:10.1038/emboj.2012.331. PMC 3567490. PMID 23241950.
- Zeng Y, Chen T (March 2019). "DNA Methylation Reprogramming during Mammalian Development". Genes (Basel). 10 (4): 257. doi:10.3390/genes10040257. PMC 6523607. PMID 30934924.
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Garcia Vizcaino JC, Schuetz AL, Burkhardt S, Benito E, Navarro Sala M, Javan SB, Haass C, Schmid B, Fischer A, Bonn S (January 2016). "DNA methylation changes in plasticity genes accompany the formation and maintenance of memory". Nat. Neurosci. 19 (1): 102–10. doi:10.1038/nn.4194. PMC 4700510. PMID 26656643.
- Kim JJ, Jung MW (2006). "Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review". Neurosci Biobehav Rev. 30 (2): 188–202. doi:10.1016/j.neubiorev.2005.06.005. PMC 4342048. PMID 16120461.
- Edwards JR, O'Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, Gingrich JA, Haghighi F, Nutter R, Bestor TH (July 2010). "Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns". Genome Res. 20 (7): 972–80. doi:10.1101/gr.101535.109. PMC 2892098. PMID 20488932.
- Pfeifer GP (April 2018). "Defining Driver DNA Methylation Changes in Human Cancer". Int J Mol Sci. 19 (4): 1166. doi:10.3390/ijms19041166. PMC 5979276. PMID 29649096.
- Veland N, Hardikar S, Zhong Y, Gayatri S, Dan J, Strahl BD, Rothbart SB, Bedford MT, Chen T (December 2017). "The Arginine Methyltransferase PRMT6 Regulates DNA Methylation and Contributes to Global DNA Hypomethylation in Cancer". Cell Rep. 21 (12): 3390–3397. doi:10.1016/j.celrep.2017.11.082. PMC 5753604. PMID 29262320.
- Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, Kelly JD, Neal DE, Maehara Y, Ponder BA, Nakamura Y, Hamamoto R (February 2011). "Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers". Int. J. Cancer. 128 (3): 562–73. doi:10.1002/ijc.25366. PMID 20473859.
- Bayraktar G, Kreutz MR (2018). "The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders". Frontiers in Molecular Neuroscience. 11: 169. doi:10.3389/fnmol.2018.00169. PMC 5975432. PMID 29875631.
- Bayraktar G, Kreutz MR (2018). "The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders". Front Mol Neurosci. 11: 169. doi:10.3389/fnmol.2018.00169. PMC 5975432. PMID 29875631.
- Jin SG, Zhang ZM, Dunwell TL, Harter MR, Wu X, Johnson J, Li Z, Liu J, Szabó PE, Lu Q, Xu GL, Song J, Pfeifer GP (January 2016). "Tet3 Reads 5-Carboxylcytosine through Its CXXC Domain and Is a Potential Guardian against Neurodegeneration". Cell Rep. 14 (3): 493–505. doi:10.1016/j.celrep.2015.12.044. PMC 4731272. PMID 26774490.
- Melamed P, Yosefzon Y, David C, Tsukerman A, Pnueli L (2018). "Tet Enzymes, Variants, and Differential Effects on Function". Front Cell Dev Biol. 6: 22. doi:10.3389/fcell.2018.00022. PMC 5844914. PMID 29556496.
- Zhang W, Xia W, Wang Q, Towers AJ, Chen J, Gao R, Zhang Y, Yen CA, Lee AY, Li Y, Zhou C, Liu K, Zhang J, Gu TP, Chen X, Chang Z, Leung D, Gao S, Jiang YH, Xie W (December 2016). "Isoform Switch of TET1 Regulates DNA Demethylation and Mouse Development". Mol. Cell. 64 (6): 1062–1073. doi:10.1016/j.molcel.2016.10.030. PMID 27916660.
- Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, Shih AH, Levine RL, Bernard O, Mercher T, Solary E, Urh M, Daniels DL, Fuks F (March 2013). "TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS". EMBO J. 32 (5): 645–55. doi:10.1038/emboj.2012.357. PMC 3590984. PMID 23353889.
- Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, Pan F, Zhao J, Hu Z, Sekhar C, Guo Z (September 2016). "OGG1 is essential in oxidative stress induced DNA demethylation". Cell. Signal. 28 (9): 1163–71. doi:10.1016/j.cellsig.2016.05.021. PMID 27251462.
- Sun Z, Xu X, He J, Murray A, Sun MA, Wei X, Wang X, McCoig E, Xie E, Jiang X, Li L, Zhu J, Chen J, Morozov A, Pickrell AM, Theus MH, Xie H (August 2019). "EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity". Nat Commun. 10 (1): 3892. Bibcode:2019NatCo..10.3892S. doi:10.1038/s41467-019-11905-3. PMC 6715719. PMID 31467272.
- Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS (April 2006). "A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA". Proc. Natl. Acad. Sci. U.S.A. 103 (15): 5752–7. Bibcode:2006PNAS..103.5752B. doi:10.1073/pnas.0509723103. PMC 1458645. PMID 16585517.
- Abdou I, Poirier GG, Hendzel MJ, Weinfeld M (January 2015). "DNA ligase III acts as a DNA strand break sensor in the cellular orchestration of DNA strand break repair". Nucleic Acids Res. 43 (2): 875–92. doi:10.1093/nar/gku1307. PMC 4333375. PMID 25539916.
- van der Kemp PA, Charbonnier JB, Audebert M, Boiteux S (2004). "Catalytic and DNA-binding properties of the human Ogg1 DNA N-glycosylase/AP lyase: biochemical exploration of H270, Q315 and F319, three amino acids of the 8-oxoguanine-binding pocket". Nucleic Acids Res. 32 (2): 570–8. doi:10.1093/nar/gkh224. PMC 373348. PMID 14752045.
- Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A (September 2004). "In situ analysis of repair processes for oxidative DNA damage in mammalian cells". Proc. Natl. Acad. Sci. U.S.A. 101 (38): 13738–43. Bibcode:2004PNAS..10113738L. doi:10.1073/pnas.0406048101. PMC 518826. PMID 15365186.
- Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A (2001). "A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA". Nucleic Acids Res. 29 (10): 2117–26. doi:10.1093/nar/29.10.2117. PMC 55450. PMID 11353081.
- Ming X, Matter B, Song M, Veliath E, Shanley R, Jones R, Tretyakova N (March 2014). "Mapping structurally defined guanine oxidation products along DNA duplexes: influence of local sequence context and endogenous cytosine methylation". J. Am. Chem. Soc. 136 (11): 4223–35. doi:10.1021/ja411636j. PMC 3985951. PMID 24571128.
- Sun Z, Xu X, He J, Murray A, Sun MA, Wei X, Wang X, McCoig E, Xie E, Jiang X, Li L, Zhu J, Chen J, Morozov A, Pickrell AM, Theus MH, Xie H (August 2019). "EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity". Nat Commun. 10 (1): 3892. Bibcode:2019NatCo..10.3892S. doi:10.1038/s41467-019-11905-3. PMC 6715719. PMID 31467272.
- Duclot F, Kabbaj M (2017). "The Role of Early Growth Response 1 (EGR1) in Brain Plasticity and Neuropsychiatric Disorders". Front Behav Neurosci. 11: 35. doi:10.3389/fnbeh.2017.00035. PMC 5337695. PMID 28321184.
- Uribe-Lewis S, Carroll T, Menon S, Nicholson A, Manasterski PJ, Winton DJ, Buczacki SJ, Murrell A (January 2020). "5-hydroxymethylcytosine and gene activity in mouse intestinal differentiation". Sci Rep. 10 (1): 546. Bibcode:2020NatSR..10..546U. doi:10.1038/s41598-019-57214-z. PMC 6969059. PMID 31953501.
- Wu X, Li G, Xie R (October 2018). "Decoding the role of TET family dioxygenases in lineage specification". Epigenetics Chromatin. 11 (1): 58. doi:10.1186/s13072-018-0228-7. PMC 6172806. PMID 30290828.
- Stremenova Spegarova, Jarmila; Lawless, Dylan; Mohamad, Siti Mardhiana Binti; Engelhardt, Karin Regine; Doody, Gina M; Shrimpton, Jennifer; Rensing-Ehl, Anne; Ehl, Stephan; Rieux-Laucat, Frederic; Cargo, Catherine; Griffin, Helen (2020-06-09). "Germline TET2 Loss-Of-Function Causes Childhood Immunodeficiency And Lymphoma". Blood: blood.2020005844. doi:10.1182/blood.2020005844. ISSN 0006-4971.
- Levin, Joshua D; Demple, Bruce (1990). "Analysis of class II (hydrolytic) and class I (beta-lyase) apurinic/apyrimidinic endonucleases with a synthetic DNA substrate". Nucleic Acids Research. 18 (17): 5069–75. doi:10.1093/nar/18.17.5069. PMC 332125. PMID 1698278.
- Karpova NN (January 2014). "Role of BDNF epigenetics in activity-dependent neuronal plasticity". Neuropharmacology. 76 Pt C: 709–18. doi:10.1016/j.neuropharm.2013.04.002. PMID 23587647.
- Fernandes J, Arida RM, Gomez-Pinilla F (September 2017). "Physical exercise as an epigenetic modulator of brain plasticity and cognition". Neurosci Biobehav Rev. 80: 443–456. doi:10.1016/j.neubiorev.2017.06.012. PMC 5705447. PMID 28666827.
- Louwies T (2018). "DNA hypomethylation in association with internal and external markers of traffic exposure in a panel of healthy adults". Air Quality, Atmosphere & Health. 11 (6): 673–681. doi:10.1007/s11869-018-0574-4.
- Weng YL, An R, Cassin J, Joseph J, Mi R, Wang C, Zhong C, Jin SG, Pfeifer GP, Bellacosa A, Dong X, Hoke A, He Z, Song H, Ming GL (April 2017). "An Intrinsic Epigenetic Barrier for Functional Axon Regeneration". Neuron. 94 (2): 337–346.e6. doi:10.1016/j.neuron.2017.03.034. PMC 6007997. PMID 28426967.
- Loh YE, Koemeter-Cox A, Finelli MJ, Shen L, Friedel RH, Zou H (February 2017). "Comprehensive mapping of 5-hydroxymethylcytosine epigenetic dynamics in axon regeneration". Epigenetics. 12 (2): 77–92. doi:10.1080/15592294.2016.1264560. PMC 5330438. PMID 27918235.