Dysosteosclerosis
Dysosteosclerosis (DSS), also known as autosomal recessive dysosteosclerosis or X-linked recessive dysosteosclerosis,[1] is a rare osteoclast-poor form of osteosclerosis that is presented during infancy and early childhood, characterized by progressive osteosclerosis and platyspondyly.[2][3] Platyspondyly and other skeletal abnormalities are radiographic features of the disease which distinguish DSS from other osteosclerotic disorders. Patients usually suffer from neurological and psychological deterioration, therefore patients are commonly associated with delayed milestones.
The cause of DSS is unclear. Different genetic mutations are observed in patients, therefore it is suggested that the cause is genetically heterogeneous. Genetic mutations responsible include, but are not limited to, TCIRG1, TNFRSF11A , and SLC29A3. It is congenital and inherited as an autosomal recessive disorder, however, an X-linked recessive inheritance is outlined in some families.[4] There is no cure for DSS. Supportive care includes orthopaedic care. Symptomatic treatment involves the reduction in calcium intake in diet.[5] Less than 30 cases of DSS have been reported in literature to date.[6]
Symptoms and Signs
The main signs and symptoms of DSS includes:[1][7]
- Skeletal abnormalities: platyspondyly, sclerosis
- Physical abnormalities: short stature, macrocephaly
- Neurological abnormalities: delayed development, mental retardation
Skeletal abnormalities
In general, patients with DSS develop osteopenia and bone fragility. DSS also affects specific areas of the human skeleton, such as the spine, skull, pelvis, and limbs.
The most common sign of DSS is platyspondyly, which is the flattening of vertebral bodies of the axial skeleton, present in 80-99% of individuals with DSS. Other spinal abnormalities associated with DSS include widened intervertebral disks, small, dense vertebral bodies, irregular vertebral endplates, hypoplastic vertebral bodies, and pronounced vertebral anterior notches.[6][7]
More than 80% of patients also reported abnormalities of the skull. Common symptoms include craniofacial hyperostosis, which is the excessive growth of bone in the skull and face. Skull base sclerosis, periorbital sclerosis, hypoplastic mandibular condyle, and absent paranasal or frontal sinuses are present in rare cases.[6][7]
It is also reported that individuals may also suffer from pelvic abnormalities. This includes the development of narrow iliac wings, as well as widened femoral necks. However, incidence of both symptoms are rare in patients with DSS, occurring in less than 30% of cases.[6][7]
The occurrence of DSS also leads to deformities of the limbs. 80% of patients reported with abnormalities of the metaphysis such as metaphyseal flaring, radiolucent metaphyses, abnormal metaphyseal trabeculation, which is abnormal trabecula patterns in the metaphyseal region, and epimetaphyseal sclerosis. Other limbic abnormalities include progressive bowing of long bones, which is present in rare cases.[6][7]
Physical abnormalities
In general, patients appear to be short statured. In addition, as a result of sclerosis of the frontal and parietal region of the calvarium,[8] macrocephaly and square shaped heads are reported in more than 80% of patients.[1][4]
Abnormal skin conditions of red-violet macular mottled skins over the entire body are occasionally observed in patients. However it is unclear whether this clinical feature is relevant to diagnosis of DSS.[7][9]
Neurological abnormalities
Visual problems are often found in people with DSS. Patients experience optic atrophy due to progressive cranial nerve compression, which may lead to nystagmus or even blindness in severe cases.[4][10]
Other neurological abnormalities include mental retardation, speech and psychological deterioration. Convulsions and status epilepticus are also present in patients, however the mechanisms for the development of these features are unknown.[4] Patients also experience cranial nerve damage resulting from progressive cranial pressure.[4][9]
Pathogenesis
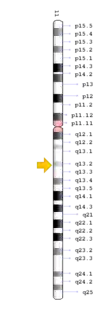
DSS is classified as an autosomal recessive disease(OMIM 224300), but it is also identified as an X-linked recessive inheritance in certain families.[4] The total number of genes that are responsible for causing the disease and the correlation between genotype and phenotype remains unclear. Multiple gene mutations were identified in different patients via whole genome sequencing, therefore it is presumed that DSS is genetically heterogeneous.
TCIRG1 mutation
The TCIRG1 gene is present in chromosome locus 11q13, which encodes for the a3 subunit of vacuolar H+ ATPase (V-ATPase) that is unique to osteoclasts.[12] The a3 subunit is responsible in anchoring the vacuolar proton pump to the ruffled membrane of osteoclasts.[9] The V-ATPase is important in mediating the transport of hydrogen ions into the resorption lacunae, which is a pit on the bone surface enclosed by the osteoclast for bone resorption. The accumulation of ions in the lacuna facilitates the decomposition of hydroxyapatite crystals by creating an acidic environment, resulting in bone resorption.[12]
Mutation of the gene results in osteoclast-rich osteopetrosis due to poor translation and altered structure of proton pump structure, which is normally involved in large amounts of osteoclast activity leading to absorption of bone tissue.[12] Mutation of TCIRG1 gene may arise from deletion or gene splicing defects, leading to frame-shifts of the nucleotides of the gene.[12]
TCIRG1 mutations illustrate the heterogeneity of DSS through a case study where DSS occurred due to a frameshift mutation, in conjunction with a mutation at an intron located in the gene in one of the alleles of chromosome 11 resulting in a splice site mutation.[9] Despite a frameshift mutation altering the C-terminal of the proton pump, due to increased remnant expression of the wild type transcript, it only resulted in intermediate autosomal recessive DSS due to the partial retainment of vacuolar proton pump function.[9]
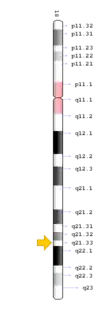
TNFRSF11A mutation
The TNFRSF11A gene is present in chromosome locus 18q21.33, which encodes for the receptor activator of NF-κB (RANK).[13] RANK is expressed in immature osteoclasts, which facilitates osteoclasts maturation upon binding of RANK ligand (RANKL). Binding of RANK ligand mediates the RANK/RANKL/OPG signalling pathway. The pathway mediates osteoclast differentiation and activation by promoting differentiation of precursors into multinucleated osteoclasts, and activating osteoclasts, thereby contributing to bone resorption and remodelling. Health conditions related to genetic changes in TNFRSF11A includes osteopetrosis, osteolysis, and Paget's disease of bone.[14][15]
Multiple reports of gene mutations exists underlining the mutation of the exons and introns leading to aberrant splicing. There are five variants to the TNFRSF11A gene which produces five unique protein iso-forms. The effects of alternative splicing on each variants are unclear. However, it is suggested that such changes lead to different expression patterns of the gene in both space and time. Some mutant splicing variants would undergo nonsense-mediated mRNA decay (NMD), while others would not be subjected to NMD, and instead produce a truncated isoform of the RANK protein. The mutated protein has structural defects hence hinders normal function in the signalling pathway, contributing to development of the disease.[14][15]
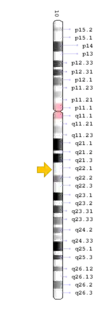
SLC29A3 mutation
The SLC29A3 gene is present in chromosome locus10q22.1, which encodes for the equilibrative nucleoside transporter 3 (ENT3), a nucleoside transporter that is present in membranes of mitochondria and lysosomes.[9] ENT3 is responsible for the trafficking of nucleoside, free purines and pyrimidines into the mitochondria and out of lysosomes.[9][16] Mutations in the gene is often accompanied by histiocytosis-lymphadenopathy plus syndrome, which is characterised by the accumulation of histiocytes leading to lymphadenopathy and other symptoms.[16]
Insertion in the coding region was reported in a patient but no records were found in any genetic databanks. Missense mutation was reported to be present in multiple patients and ranked disease-causing by MutationTaster.[9] This insertion mutation is located in the loop within transmembrane helices 1 and 2, while the missense mutations is located in the loop within transmembrane helix 6, 9, and 11.[17]
Mutation leads to impaired ENT3 transportation activity, accumulation of nucleotides and nucleoside in lysosomes.[9] As osteoclasts express the SLC29A3 gene, mutation results in incapacitated differentiated osteoclasts’ ability in demineralising calcium phosphate crystal surfaces, disrupted osteoclastic differentiation, and diminished osteoclast counts.[17] The reduced osteoclast differentiation and activity results in decreased demineralisation and reabsorption of bone structures.
Radiographic features
Whilst some symptoms of DSS can be visually identified, many key symptoms of DSS cannot be identified as such. Therefore, radiographic techniques are required to reach a correct diagnosis for DSS.
Skull
In the skull, sclerosis is predominantly observed in the cranial vault and skull base.[10] Other symptoms displayed in the skull include dental anomalies such as abnormal dentition, hypodontia and impaired tooth calcification;[9] intracranial calcifications, which is the calcification of the brain parenchyma; narrow optic canal and other cranial foramina.[18]
Abnormal features of the appendicular skeleton
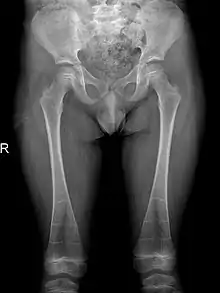
Chest
The ribs are sclerotic, truncated, expanded, and featureless. Sclerosis is also present in the sternum, clavicles and scapulae. Pectus carinatum, a chest deformity, is also characteristic of the disease.[18]
Long bones
Sclerosis of epiphyses, diaphyses, and metaphyses with increased radiolucency are key characteristics of the disease.[18] In addition, mottled metaphyseal sclerosis and widening are also present in patients.[4][9] Development of irregular patchy sclerosis along the bone can also be identified,[9] as well as metaphyseal flaring evolve towards Erlenmeyer flask deformity with nonuniform patches of sclerosis, which are especially prevalent in older patients.[10]
The metadiaphyses, a portmanteau of the metaphysis and diaphysis,[20] are bulbous and expanded with bowing and relative radiolucency. The expanded regions are also sclerotic and gives the characteristic bone-in-bone appearance.[4]
Spine
Platyspondyly is the development of flattened vertebral bodies, which is one of the most notable symptoms as it distinguishes DSS from other similar diseases such as osteosclerosis. It is observed to be most significant in the thoracic region with increased intervertebral spaces.[4] Platyspondyly development begins with generalised osteosclerosis, then subsequent interspersing sclerotic bands develop within vertebral bodies with normal bone density, showing radiolucency.[18]
Diagnosis
Medical diagnosis of DSS involves various examinations and evaluations. This usually includes physical examinations, medical history evaluations, assessment of signs and symptoms, laboratory tests and image studies. Biopsy may also be required if necessary.[1][17]
Signs and symptoms of DSS show similarities with multifarious disorders and diseases such as osteosclerosis, H syndrome and Pyle disease.[1][17] As such, it is likely that DSS is consequently incorrectly identified as osteopertrosis.[4] Therefore, additional tests may be performed in order to arrive at a definitive diagnosis.[1][17]
Prognosis and management
Overall the disease has a poor prognosis, with treatment mainly focusing on palliation and comfort care.[2]
As the mechanism and clinical course of DSS remains unclear, definitive treatment is not available for patients. Bone marrow transplant may improve skeletal abnormalities, however it is improbable the transplant will ameliorate the unexplained neurological deteriorations.[4] In addition, the surgery may not be suitable for every patients as the underlying genetic cause of the disease varies amongst patients. Advice of control and reduction in excessive calcium intake may be recommended by physicians to ensure circulating levels of parathyroid hormones are normal to induce and maintain bone resorption.[5]
References
- "Dysosteosclerosis". Dovemed.
- "Orphanet: Dysosteosclerosis". www.orpha.net.
- Penna S, Capo V, Palagano E, Sobacchi C, Villa A (2019-02-19). "One Disease, Many Genes: Implications for the Treatment of Osteopetroses". Frontiers in Endocrinology. 10: 85. doi:10.3389/fendo.2019.00085. PMC 6389615. PMID 30837952.
- Elçioglu NH, Vellodi A, Hall CM (August 2002). "Dysosteosclerosis: a report of three new cases and evolution of the radiological findings". Journal of Medical Genetics. 39 (8): 603–7. doi:10.1136/jmg.39.8.603. PMC 1735202. PMID 12161605.
- Whyte MP, Wenkert D, McAlister WH, Novack DV, Nenninger AR, Zhang X, et al. (November 2010). "Dysosteosclerosis presents as an "osteoclast-poor" form of osteopetrosis: comprehensive investigation of a 3-year-old girl and literature review". Journal of Bone and Mineral Research. 25 (11): 2527–39. doi:10.1002/jbmr.131. PMC 3179286. PMID 20499338.
- "Dysosteosclerosis". Genetic and Rare Diseases Information Center. National Institutes of Health.
- Przylepa KA. "DYSOSTEOSCLEROSIS". omim.org.
- Beighton P, Cremin BJ (6 December 2012). Sclerosing Bone Dysplasias. Springer London. pp. 95–100. ISBN 978-1-4471-1292-1.
- Howaldt A, Nampoothiri S, Quell LM, Ozden A, Fischer-Zirnsak B, Collet C, et al. (March 2019). "Sclerosing bone dysplasias with hallmarks of dysosteosclerosis in four patients carrying mutations in SLC29A3 and TCIRG1". Bone. 120: 495–503. doi:10.1016/j.bone.2018.12.002. PMID 30537558.
- John E, Kozlowski K, Masel J, Muralinath S, Vijayalakshmi G (August 1996). "Dysosteosclerosis". Australasian Radiology. 40 (3): 345–7. doi:10.1111/j.1440-1673.1996.tb00417.x. PMID 8826749.
- "TCIRG1 gene". Genetics Home Reference. Retrieved 2020-04-08.
- Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, et al. (July 2000). "Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis". Nature Genetics. 25 (3): 343–6. doi:10.1038/77131. PMID 10888887. S2CID 21316081.
- "TNFRSF11A gene". Genetics Home Reference. Retrieved 2020-04-04.
- Guo L, Elcioglu NH, Karalar OK, Topkar MO, Wang Z, Sakamoto Y, et al. (June 2018). "Dysosteosclerosis is also caused by TNFRSF11A mutation". Journal of Human Genetics. 63 (6): 769–774. doi:10.1038/s10038-018-0447-6. PMID 29568001. S2CID 4133369.
- Xue JY, Wang Z, Shinagawa S, Ohashi H, Otomo N, Elcioglu NH, et al. (October 2019). "TNFRSF11A-Associated Dysosteosclerosis: A Report of the Second Case and Characterization of the Phenotypic Spectrum". Journal of Bone and Mineral Research. 34 (10): 1873–1879. doi:10.1002/jbmr.3805. PMID 31163101.
- "SLC29A3 gene". Genetics Home Reference. National Institutes of Health.
- Campeau PM, Lu JT, Sule G, Jiang MM, Bae Y, Madan S, et al. (November 2012). "Whole-exome sequencing identifies mutations in the nucleoside transporter gene SLC29A3 in dysosteosclerosis, a form of osteopetrosis". Human Molecular Genetics. 21 (22): 4904–9. doi:10.1093/hmg/dds326. PMC 3607481. PMID 22875837.
- Castriota Scanderbeg A (26 October 2005). Abnormal Skeletal Phenotypes: From Simple Signs to Complex Diagnoses. Berlin, Heidelberg: Springer Berlin Heidelberg. pp. 690–691. ISBN 9783540679974.
- Nasman A. "Erlenmeyer flask deformity | Radiology Case | Radiopaedia.org". Radiopaedia. Retrieved 2020-04-19.
- Hacking C, Bell DJ. "Metadiaphysis". Radiopaedia.