Ligation (molecular biology)
In molecular biology, ligation is the joining of two nucleic acid fragments through the action of an enzyme. It is an essential laboratory procedure in the molecular cloning of DNA whereby DNA fragments are joined together to create recombinant DNA molecules, such as when a foreign DNA fragment is inserted into a plasmid. The ends of DNA fragments are joined together by the formation of phosphodiester bonds between the 3'-hydroxyl of one DNA terminus with the 5'-phosphoryl of another. RNA may also be ligated similarly. A co-factor is generally involved in the reaction, and this is usually ATP or NAD+.
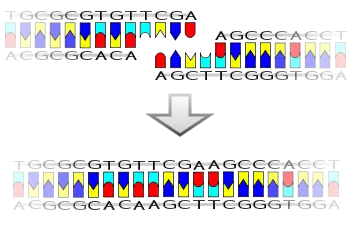
Ligation in the laboratory is normally performed using T4 DNA ligase, however, procedures for ligation without the use of standard DNA ligase are also popular.
Ligation reaction
The mechanism of the ligation reaction was first elucidated in the laboratory of I. Robert Lehman.[1][2] Two fragments of DNA may be joined together by DNA ligase which catalyzes the formation of a phosphodiester bond between the 3'-OH at one end of a strand of DNA and the 5'-phosphate group of another. In animals and bacteriophage, ATP is used as the energy source for the ligation, while In bacteria, NAD+ is used.[3]
The DNA ligase first reacts with ATP or NAD+, forming a ligase-AMP intermediate with the AMP linked to the ε-amino group of lysine in the active site of the ligase via a phosphoamide bond. This adenylyl group is then transferred to the phosphate group at the 5' end of a DNA chain, forming a DNA-adenylate complex. Finally, a phosphodiester bond between the two DNA ends is formed via the nucleophilic attack of the 3'-hydroxyl at the end of a DNA strand on the activated 5′-phosphoryl group of another.[1]
A nick in the DNA (i.e. a break in one strand of a double-stranded DNA) can be repaired very efficiently by the ligase. However, a complicating feature of ligation presents itself when ligating two separate DNA ends as the two ends need to come together before the ligation reaction can proceed. In the ligation of DNA with sticky or cohesive ends, the protruding strands of DNA may be annealed together already, therefore it is a relatively efficient process as it is equivalent to repairing two nicks in the DNA. However, in the ligation of blunt-ends, which lack protruding ends for the DNA to anneal together, the process is dependent on random collision for the ends to align together before they can be ligated, and is consequently a much less efficient process.[4] The DNA ligase from E. coli cannot ligate blunt-ended DNA except under conditions of molecular crowding, and it is therefore not normally used for ligation in the laboratory. Instead the DNA ligase from phage T4 is used as it can ligate blunt-ended DNA as well as single-stranded DNA.[5][3]
Factors affecting ligation
Factors that affect an enzyme-mediated chemical reaction would naturally affect a ligation reaction, such as the concentration of enzyme and the reactants, as well as the temperature of reaction and the length of time of incubation. Ligation is complicated by the fact that the desired ligation products for most ligation reactions should be between two different DNA molecules and the reaction involves both inter- and intra-molecular reactions, and that an additional annealing step is necessary for efficient ligation.
The three steps to form a new phosphodiester bond during ligation are: enzyme adenylylation, adenylyl transfer to DNA, and nick sealing. Mg(2+) is a cofactor for catalysis, therefore at high concentration of Mg(2+) the ligation efficiency is high. If the concentration of Mg(2+) is limited, the nick- sealing is the rate- limiting reaction of the process, and adenylylated DNA intermediate stays in the solution. Such adenylylation of the enzyme restrains the rebinding to the adenylylated DNA intermediate comparison of an Achilles' heel of LIG1, and represents a risk if they are not fixed.[6]
DNA concentration
The concentration of DNA can affect the rate of ligation, and whether the ligation is an inter-molecular or intra-molecular reaction. Ligation involves joining up the ends of a DNA with other ends, however, each DNA fragment has two ends, and if the ends are compatible, a DNA molecule can circularize by joining its own ends. At high DNA concentration, there is a greater chance of one end of a DNA molecule meeting the end of another DNA, thereby forming intermolecular ligation. At a lower DNA concentration, the chance that one end of a DNA molecule would meet the other end of the same molecule increases, therefore intramolecular reaction that circularizes the DNA is more likely. The transformation efficiency of linear DNA is also much lower than circular DNA, and for the DNA to circularize, the DNA concentration should not be too high. As a general rule, the total DNA concentration should be less than 10 μg/ml.[7]
The relative concentration of the DNA fragments, their length, as well as buffer conditions are also factors that can affect whether intermolecular or intramolecular reactions are favored.
The concentration of DNA can be artificially increased by adding condensing agents such as cobalt hexamine and biogenic polyamines such as spermidine, or by using crowding agents such as polyethylene glycol (PEG) which also increase the effective concentration of enzymes.[8][9] Note however that additives such as cobalt hexamine can produce exclusively intermolecular reaction,[10] resulting in linear concatemers rather than the circular DNA more suitable for transformation of plasmid DNA, and is therefore undesirable for plasmid ligation. If it is necessary to use additives in plasmid ligation, the use of PEG is preferable as it can promote intramolecular as well as intermolecular ligation.[11]
Ligase concentration
The higher the ligase concentration, the faster the rate of ligation. Blunt-end ligation is much less efficient than sticky end ligation, so a higher concentration of ligase is used in blunt-end ligations. High DNA ligase concentration may be used in conjunction with PEG for a faster ligation, and they are the components often found in commercial kits designed for rapid ligation.[12][13]
Temperature
Two issues are involved when considering the temperature of a ligation reaction. First, the optimum temperature for DNA ligase activity which is 37°C, and second, the melting temperature (Tm) of the DNA ends to be ligated. The melting temperature is dependent on length and base composition of the DNA overhang—the greater the number of G and C, the higher the Tm since there are three hydrogen bonds formed between G-C base pair compared to two for A-T base pair—with some contribution from the stacking of the bases between fragments. For the ligation reaction to proceed efficiently, the ends should be stably annealed, and in ligation experiments, the Tm of the DNA ends is generally much lower than 37°C. The optimal temperature for ligating cohesive ends is therefore a compromise between the best temperature for DNA ligase activity and the Tm where the ends can associate.[14] However, different restriction enzymes generates different ends, and the base composition of the ends produced by these enzymes may also differ, the melting temperature and therefore the optimal temperature can vary widely depending on the restriction enzymes used, and the optimum temperature for ligation may be between 4-15°C depending on the ends.[15][16] Ligations also often involve ligating ends generated from different restriction enzymes in the same reaction mixture, therefore it may not be practical to select optimal temperature for a particular ligation reaction and most protocols simply choose 12-16°C, room temperature, or 4°C.
Buffer composition
The ionic strength of the buffer used can affect the ligation. The kinds of cations presence can also influence the ligation reaction, for example, excess amount of Na+ can cause the DNA to become more rigid and increase the likelihood of intermolecular ligation. At high concentration of monovalent cation (>200 mM) ligation can also be almost completely inhibited.[17] The standard buffer used for ligation is designed to minimize ionic effects.[18]
Sticky-end ligation
Restriction enzymes can generate a wide variety of ends in the DNA they digest, but in cloning experiments most commonly-used restriction enzymes generate a 4-base single-stranded overhang called the sticky or cohesive end (exceptions include NdeI which generates a 2-base overhang, and those that generate blunt ends). These sticky ends can anneal to other compatible ends and become ligated in a sticky-end (or cohesive end) ligation. EcoRI for example generates an AATT end, and since A and T have lower melting temperature than C and G, its melting temperature Tm is low at around 6°C.[19] For most restriction enzymes, the overhangs generated have a Tm that is around 15°C.[18] For practical purposes, sticky end ligations are performed at 12-16°C, or at room temperature, or alternatively at 4°C for a longer period.
For the insertion of a DNA fragment into a plasmid vector, it is preferable to use two different restriction enzymes to digest the DNA so that different ends are generated. The two different ends can prevent the religation of the vector without any insert, and it also allows the fragment to be inserted in a directional manner.
When it is not possible to use two different sites, then the vector DNA may need to be dephosphorylated to avoid a high background of recircularized vector DNA with no insert. Without a phosphate group at the ends the vector cannot ligate to itself, but can be ligated to an insert with a phosphate group. Dephosphorylation is commonly done using calf-intestinal alkaline phosphatase (CIAP) which removes the phosphate group from the 5′ end of digested DNA, but note that CIAP is not easy to inactivate and can interfere with ligation without an additional step to remove the CIAP, thereby resulting in failure of ligation. CIAP should not be used in excessive amount and should only be used when necessary. Shrimp alkaline phosphatase (SAP) or Antarctic phosphatase (AP) are suitable alternative as they can be easily inactivated.
Blunt-end ligation
Blunt end ligation does not involve base-pairing of the protruding ends, so any blunt end may be ligated to another blunt end. Blunt ends may be generated by restriction enzymes such as SmaI and EcoRV. A major advantage of blunt-end cloning is that the desired insert does not require any restriction sites in its sequence as blunt-ends are usually generated in a PCR, and the PCR generated blunt-ended DNA fragment may then be ligated into a blunt-ended vector generated from restriction digest.
Blunt-end ligation, however, is much less efficient than sticky end ligation, typically the reaction is 100X slower than sticky-end ligation. Since blunt-end does not have protruding ends, the ligation reaction depends on random collisions between the blunt-ends and is consequently much less efficient. To compensate for the lower efficiency, the concentration of ligase used is higher than sticky end ligation (10x or more). The concentration of DNA used in blunt-end ligation is also higher to increase the likelihood of collisions between ends, and longer incubation time may also be used for blunt-end ligations.
If both ends needed to be ligated into a vector are blunt-ended, then the vector needs to be dephosphorylated to minimize self-ligation. This may be done using CIAP, but caution in its use is necessary as noted previously. Since the vector has been dephosphorylated, and ligation requires the presence of a 5'-phosphate, the insert must be phosphorylated. Blunt-ended PCR product normally lacks a 5'-phosphate, therefore it needs to be phosphorylated by treatment with T4 polynucleotide kinase.[20]
Blunt-end ligation is also reversibly inhibited by high concentration of ATP.[21]
PCR usually generates blunt-ended PCR products, but note that PCR using Taq polymerase can add an extra adenine (A) to the 3' end of the PCR product. This property may be exploited in TA cloning where the ends of the PCR product can anneal to the T end of a vector. TA ligation is therefore a form of sticky end ligation. Blunt-ended vectors may be turned into vector for TA ligation with dideoxythymidine triphosphate (ddTTP) using terminal transferase.
General guidelines
For the cloning of an insert into a circular plasmid:
- The total DNA concentration used should be less than 10 μg/ml as the plasmid needs to recircularize.
- The molar ratio of insert to vector is usually used at around 3:1. Very high ratio may produce multiple inserts. The ratio may be adjusted depending on the size of the insert, and other ratios may be used, such as 1:1.
Trouble-shooting
Sometimes ligation fail to produce the desired ligated products, and some of the possible reasons may be:
- Damaged DNA – Over-exposure to UV radiation during preparation of DNA for ligation can damage the DNA and significantly reduce transformation efficiency. A higher-wavelength UV radiation (365 nm) which cause less damage to DNA should be used if it is necessary work for work on the DNA on a UV transilluminator for an extended period of time. Addition of cytidine or guanosine to the electrophoresis buffer at 1 mM concentration however may protect the DNA from damage.[22]
- Incorrect usage of CIAP or its inefficient inactivation or removal.
- Excessive amount of DNA used.
- Incomplete DNA digest – The vector DNA that is incompletely digested will give rise to a high background, and this may be checked by doing a ligation without insert as a control. Insert that is not completely digested will also not ligate properly and circularize. When digesting a PCR product, make sure that sufficient extra bases have been added to the 5'-ends of the oligonucleotides used for PCR as many restriction enzymes require a minimum number of extra basepairs for efficient digest. The information on the minimum basepair required is available from restriction enzyme suppliers such as in the catalog of New England Biolabs.[23]
- Incomplete ligation – Blunt-ends DNA (e.g. SmaI) and some sticky-ends DNA (e.g. NdeI) that have low-melting temperature require more ligase and longer incubation time.[24]
- Protein expressed from ligated gene insert is toxic to cells.
- Homologous sequence in insert to sequence in plasmid DNA resulting in deletion.
Other methods of DNA ligation
A number of commercially available DNA cloning kits use other methods of ligation that do not require the use of the usual DNA ligases. These methods allow cloning to be done much more rapidly, as well as allowing for simpler transfer of cloned DNA insert to different vectors. These methods however require the use of specially designed vectors and components, and may lack flexibility.
Topoisomerase-mediated ligation
Topoisomerase can be used instead of ligase for ligation, and the cloning may be done more rapidly without the need for restriction digest of the vector or insert. In this TOPO cloning method a linearized vector is activated by attaching topoisomerase I to its ends, and this "TOPO-activated" vector may then accept a PCR product by ligating to both of the 5' ends of the PCR product, the topoisomerase is released and a circular vector is formed in the process.[25]
Homologous recombination
Another method of cloning without the use of ligase is by DNA recombination, for example as used in the Gateway cloning system.[26][27] The gene, once cloned into the cloning vector (called entry clone in this method), may be conveniently introduced into a variety of expression vectors by recombination.[28]
See also
References
- Nicole Kresge, Robert D. Simoni and Robert L. Hill (January 12, 2007). "Insights into DNA Joining: I. Robert Lehman's Work on DNA Ligase". The Journal of Biological Chemistry. 282.CS1 maint: uses authors parameter (link)
- Modorich P, Lehman IR (10 November 1973). "Deoxyribonucleic acid ligase. A steady state kinetic analysis of the reaction catalyzed by the enzyme from Escherichia coli" (PDF). J Biol Chem. 248 (21): 7502–11. PMID 4355585.
- Lubert Stryer (2007). Biochemistry (3rd ed.). Freeman. pp. 658–659.
- Venetia A. Saunders (6 December 2012). Microbial genetics applied to biotechnology :: principles and techniques of Gene Transfer and Manipulation. Springer. ISBN 9781461597964.
- Robert W. Old; Sandy B. Primrose (1994-09-27). Principle of Gene Manipulation - An Introduction to Genetic Engineering (5th ed.). Blackwell Scientific. p. 37. ISBN 9780632037124.
- Taylor, Conrad,Wahl, O'brian (July 2011). "Kinetic mechanism of human DNA ligase I reveals magnesium-dependent changes in the rate-limiting step that compromise ligation efficiency". The Journal of Biological Chemistry. 286 (26): 23054–23062. doi:10.1074/jbc.m111.248831. PMC 3123073. PMID 21561855 – via one search.CS1 maint: multiple names: authors list (link)
- Joseph Sambrook; David Russell. "Chapter 1: Plasmids and Their Usefulness in Molecular Cloning". Molecular Cloning - A Laboratory Manual. 1 (3rd ed.). pp. 1.20–1.21. ISBN 978-0-87969-577-4.
- J R Rusche; P Howard-Flanders (March 25, 1985). "Hexamine cobalt chloride promotes intermolecular ligation of blunt end DNA fragments by T4 DNA ligase". Nucleic Acids Research. 13 (6): 1997–2008. doi:10.1093/nar/13.6.1997. PMC 341130. PMID 4000951.
- Zimmerman SB, Pheiffer BH (1983). "Macromolecular crowding allows blunt-end ligation by DNA ligases from rat liver or Escherichia coli". Proceedings of the National Academy of Sciences of the United States of America. 80 (19): 5852–6. doi:10.1073/pnas.80.19.5852. PMC 390173. PMID 6351067.
- James R. Rusche; Paul Howard-Flanders (Mar 25, 1985). "Hexamine cobalt chloride promotes intermolecular ligation of blunt end DNA fragments by T4 DNA ligase". Nucleic Acids Research. 13 (6): 1997–2008. doi:10.1093/nar/13.6.1997. PMC 341130. PMID 4000951.
- K Hayashi; M Nakazawa; Y Ishizaki; N Hiraoka; A Obayashi (October 10, 1986). "Regulation of inter- and intramolecular ligation with T4 DNA ligase in the presence of polyethylene glycol". Nucleic Acids Research. 14 (19): 7617–7631. doi:10.1093/nar/14.19.7617. PMC 311784. PMID 3022231.
- "FAQ". NEB.
- "Quick Ligation Kit". NEB.
- Robert W. Old; Sandy B. Primrose (1994-09-27). Principle of Gene Manipulation - An Introduction to Genetic Engineering (5th ed.). Blackwell Scientific. p. 36. ISBN 9780632037124.
- L Ferretti; V Sgaramella (January 10, 1981). "Temperature dependence of the joining by T4 DNA ligase of termini produced by type II restriction endonucleases". Nucleic Acids Research. 9 (1): 85–93. doi:10.1093/nar/9.1.85. PMC 326670. PMID 6259621.
- Dugaiczyk A, Boyer HW, Goodman HM (July 25, 1975). "Ligation of EcoRI endonuclease-generated DNA fragments into linear and circular structures". Journal of Molecular Biology. 96 (1): 171–84. doi:10.1016/0022-2836(75)90189-8. PMID 169355.
- Raae AJ, Kleppe RK, Kleppe K (December 15, 1975). "Kinetics and effect of salts and polyamines on T4 polynucleotide ligase". European Journal of Biochemistry. 60 (2): 437–43. doi:10.1111/j.1432-1033.1975.tb21021.x. PMID 173544.
- G. Lucotte; F. Baneyx (1993). Introduction to Molecular Cloning Techniques. Wiley-Blackwell. p. 156. ISBN 978-0471188490.
- Janet E. Mertz; Ronald W. Davis (1972). "Cleavage of DNA by R1 Restriction Endonuclease Generates Cohesive Ends". Proceedings of the National Academy of Sciences of the United States of America. 69 (11): 3370–3374. doi:10.1073/pnas.69.11.3370. PMC 389773. PMID 4343968.
- "Cloning Guide". New England Biolabs Inc.
- L Ferretti; V Sgaramella (August 11, 1981). "Specific and reversible inhibition of the blunt end joining activity of the T4 DNA ligase". Nucleic Acids Research. 9 (15): 3695–3705. doi:10.1093/nar/9.15.3695. PMC 327385. PMID 6269089.
- Gründemann D, Schömig E (1996). "Protection of DNA during preparative agarose gel electrophoresis against damage induced by ultraviolet light" (PDF). BioTechniques. 21 (5): 898–903. doi:10.2144/96215rr02. PMID 8922632.
- "Cleavage Close to the End of DNA Fragments". New England Biolabs Inc.
- Chang E.; Ge B.; Lee M.; So M.; Wang W. (2005). "Investigation of the Ligation Efficiency of NdeI Digested Fragments" (PDF). Journal of Experimental Microbiology and Immunology. 7: 68–72. Retrieved 2012-10-29. (Note that this paper has an error describing NdeI as a four-base cutter.)
- "The Technology Behind TOPO® Cloning". Invitrogen.
- Esposito D, Garvey LA, Chakiath CS (2009). "Gateway cloning for protein expression". High Throughput Protein Expression and Purification. Methods in Molecular Biology. 498. pp. 31–54. doi:10.1007/978-1-59745-196-3_3. ISBN 978-1-58829-879-9. PMID 18988017.
- "Cloning Methods - Recombination cloning systems". EMBL.
- "Gateway® Recombination Cloning Technology". Invitrogen.