Light sheet fluorescence microscopy
Light sheet fluorescence microscopy (LSFM) is a fluorescence microscopy technique with an intermediate-to-high[1] optical resolution, but good optical sectioning capabilities and high speed. In contrast to epifluorescence microscopy only a thin slice (usually a few hundred nanometers to a few micrometers) of the sample is illuminated perpendicularly to the direction of observation. For illumination, a laser light-sheet is used, i.e. a laser beam which is focused only in one direction (e.g. using a cylindrical lens). A second method uses a circular beam scanned in one direction to create the lightsheet. As only the actually observed section is illuminated, this method reduces the photodamage and stress induced on a living sample. Also the good optical sectioning capability reduces the background signal and thus creates images with higher contrast, comparable to confocal microscopy. Because LSFM scans samples by using a plane of light instead of a point (as in confocal microscopy), it can acquire images at speeds 100 to 1000 times faster than those offered by point-scanning methods.


This method is used in cell biology[2] and for microscopy of intact, often chemically cleared, organs, embryos, and organisms.[3]
Starting in 1994, LSFM was developed as orthogonal plane fluorescence optical sectioning microscopy or tomography (OPFOS)[4] mainly for large samples and later as the selective/single plane illumination microscopy (SPIM) also with sub-cellular resolution.[5] This introduced an illumination scheme into fluorescence microscopy, which has already been used successfully for dark field microscopy under the name ultramicroscopy.[6]
Setup of LSFM
Basic setup

In this type of microscopy,[7] the illumination is done perpendicularly to the direction of observation (see schematic image at the top of the article). The expanded beam of a laser is focused in only one direction by a cylindrical lens, or by a combination of a cylindrical lens and a microscope objective as the latter is available in better optical quality and with higher numerical aperture than the first. This way a thin sheet of light or lightsheet is created in the focal region that can be used to excite fluorescence only in a thin slice (usually a few micrometers thin) of the sample.
The fluorescence light emitted from the lightsheet is then collected perpendicularly with a standard microscope objective and projected onto an imaging sensor (usually a CCD, electron multiplying CCD or CMOS camera). In order to let enough space for the excitation optics/lightsheet an observation objective with high working distance is used. In most LSFMs the detection objective and sometimes also the excitation objective are fully immersed in the sample buffer, so usually the sample and excitation/detection optics are embedded into a buffer-filled sample chamber, which can also be used to control the environmental conditions (temperature, carbon dioxide level ...) during the measurement. The sample mounting in LSFM is described below in more detail.
As both the excitation lightsheet and the focal plane of the detection optics have to coincide to form an image, focusing different parts of the sample can not be done by translating the detection objective, but usually the whole sample is translated and rotated instead.
Extensions of the basic LSFM idea
In recent years, several extensions to this scheme have been developed:
- The use of two counter-propagating lightsheets helps to reduce typical SPIM artifacts, like shadowing (see first z-stack above)[8]
- In addition to counter-propagating lightsheets a setup with detection from two opposing sides has been proposed in 2012.[9][10] This allows measurement of z- and rotation-stacks for a full 3D reconstruction of the sample more rapidly.
- The lightsheet can also be created by scanning a normal laser focus up and down.[11] This also allows use of self-reconstructing beams (such as bessel beams or Airy beams) for the illumination which improve the penetration of the lightsheet into thick samples, as the negative effect of scattering on the lightsheet is reduced.[12][13][14] These self-reconstructing beams can be modified to counteract intensity losses using attenuation-compensation techniques, further increasing the signal collected from within thick samples.[15]
- In oblique plane microscopy (OPM)[16] the detection objective is used to also create the lightsheet: The lightsheet is now emitted from this objective under an angle of about 60°. Additional optics is used to also tilt the focal plane used for detection by the same angle.
- LSFM has also been combined with two-photon (2P) excitation, which improves the penetration into thick and scattering samples.[17] Use of 2P excitation in near-infrared wavelengths has been used to replace 1P excitation in blue-visible wavelengths in brain imaging experiments involving response to visual stimuli.[18]
- SPIM can also be combined with techniques such as fluorescence correlation spectroscopy, to allow spatially resolved mobility measurements of fluorescing particles (e.g. fluorescent beads, quantum dots or fluorescently labeled proteins) inside living biological samples.[19][20]
- Also a combination of a SPIM microscope with a gated image intensifier camera has been reported that allowed measuring a map of fluorescence lifetimes (fluorescence lifetime imaging, FLIM).[21]
- LSFM was combined with super resolution microscopy techniques to improve its resolution beyond the Abbe limit.[22][23] Also a combination of stimulated emission depletion microscopy (STED) and SPIM has been published, that leads to a reduced lightsheet thickness due to the STED effect.[24] See also the section on the power of resolution of LSFM below.
- LSFM was modified to be compatible with all objectives, even coverslip-based, oil-immersion objectives with high numerical aperture to increase native spatial resolution and fluorescence detection efficiency.[25] This technique involves tilting the light sheet relative to the detection objective at a precise angle to allow the light sheet to form on the surface of glass coverslips.
- LSFM was combined with Adaptive Optics techniques in 2012 to improve the depth of imaging in thick and inhomogenous samples at a depth of 350 um.[26] A Shack Hartmann wavefront sensor was positioned in the detection path and guide stars are used in a close feedback loop. In his thesis,[27] the author discuss the advantage of having Adaptive Optics both in the illumination and detection path of the LSFM to correct aberrations induced by the sample.
Sample mounting

The separation of the illumination and detection beampaths in LSFM (except in oblique plane microscopy) creates a need for specialized sample mounting methods. To date most LSFMs are built in such a way that the illumination and detection beampath lie in a horizontal plane (see illustrations above), thus the sample is usually hanging from the top into the sample chamber or is resting on a vertical support inside the sample chamber. Several methods have been developed to mount all sorts of samples:
- Fixed (and potentially also cleared) samples can be glued to a simple support or holder and can stay in their fixing solution during imaging.
- Larger living organisms are usually sedated and mounted in a soft gel cylinder that is extruded from a (glass or plastic) capillary hanging from above into the sample chamber.
- Adherent cells can be grown on small glass plates that are hanging in the sample chamber.
- Plants can be grown in clear gels containing a growth medium. The gels are cut away at the position of imaging, so they do not reduce the lightsheet and image quality by scattering and absorption.[28]
- Liquid samples (e.g. for fluorescence correlation spectroscopy) can be mounted in small bags made of thin plastic foil matching the refractive index of the surrounding immersion medium in the sample chamber.[20]
Some LSFMs have been developed where the sample is mounted as in standard microscopy (e.g. cells grow horizontally on the bottom of a petri dish) and the excitation and detection optics are constructed in an upright plane from above. This also allows combining a LSFM with a standard inverted microscope and avoids the requirement for specialized sample mounting procedures.[19][29][30][31]
LSFM image properties
Typical imaging modes
Most LSFMs are used to produce 3D images of the sample by moving the sample through the image plane. If the sample is larger than the field of view of the image sensor, the sample also has to be shifted laterally. An alternative approach is to move the image plane through the sample to create the image stack.[31]
Long experiments can be carried out, for example with stacks recorded every 10 sec–10 min over the timespan of days. This allows study of changes over time in 3D, or so-called 4D microscopy.
After the image acquisition the different image stacks are registered to form one single 3D dataset. Multiple views of the sample can be collected, either by interchanging the roles of the objectives[31] or by rotating the sample.[8] Having multiple views can yield more information than a single stack; for example occlusion of some parts of the sample may be overcome. Multiple views also improves 3D image resolution by overcoming poor axial resolution as described below.
Some studies also use a SPIM to image only one slice of the sample, but at much higher temporal resolution. This allows e.g. to observe the beating heart of a zebra fish embryo in real-time.[32] Together with fast translation stages for the sample a high-speed 3D particle tracking has been implemented.[33]
Power of resolution
The lateral resolution of a SPIM is comparable to that of a standard (epi) fluorescence microscope, as it is determined fully by the detection objective and the wavelength of the detected light (see Abbe limit). E.g. for detection in the green spectral region around 525 nm, a resolution of 250–500 nm can be reached.[7] The axial resolution is worse than the lateral (about a factor of 4), but it can be improved by using a thinner lightsheet in which case nearly isotropic resolution is possible.[19] Thinner light sheets are either thin only in a small region (for Gaussian beams) or else specialized beam profiles such as Bessel beams must be used (besides added complexity, such schemes add side lobes which can be detrimental[13]). Alternatively, isotropic resolution can be achieved by computationally combining 3D image stacks taken from the same sample under different angles. Then the depth-resolution information lacking in one stack is supplied from another stack; for example with two orthogonal stacks the (poor-resolution) axial direction in one stack is a (high-resolution) lateral direction in the other stack.
The lateral resolution of LSFM can be improved beyond the Abbe limit, by using super resolution microscopy techniques, e.g. with using the fact, that single fluorophores can be located with much higher spatial precision than the nominal resolution of the used optical system (see stochastic localization microscopy techniques).[22] Also structured illumination techniques have been applied to further improve the optical sectioning capacity of LSFM.[23]
Stripe artifacts
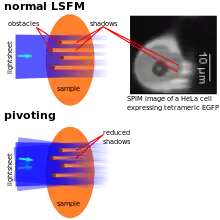
As the illumination typically penetrates the sample from one side, obstacles lying in the way of the lightsheet can disturb its quality by scattering and/or absorbing the light. This typically leads to dark and bright stripes in the images. If parts of the samples have a significantly higher refractive index (e.g. lipid vesicles in cells), they can also lead to a focussing effect resulting in bright stripes behind these structures. To overcome this artifact, the lightsheets can e.g. be "pivoting". That means that the lightsheet's direction of incidence is changed rapidly (~1 kHz rate) by a few degrees (~10°), so light also hits the regions behind the obstacles. Illumination can also be performed with two (pivoted) lightsheets (see above) to further reduce these artifacts.[8] Alternatively, an algorithm called VSNR (Variational Stationary Noise Remover) has been developed and is available as a free Fiji plugin. [34]
History
At the beginning of the 20th century, R. A. Zsigmondy introduced the ultramicroscope as a new illumination scheme into dark-field microscopy. Here sunlight or a white lamp is used to illuminate a precision slit. The slit is then imaged by a condensor lens into the sample to form a lightsheet. Scattering (sub-diffractive) particles can be observed perpendicularly with a microscope. This setup allowed the observation of particles with sizes smaller than the microscope's resolution and led to a Nobel prize for Zsigmondy in 1925.[35]
The first application of this illumination scheme for fluorescence microscopy was published in 1993 by Voie et al. under the name orthogonal-plane fluorescence optical sectioning (OPFOS).[4] for imaging of the internal structure of the cochlea. The resolution at that time was limited to 10 µm laterally and 26 µm longitudinally but at a sample size in the millimeter range. The OPFOS microscope used a simple cylindrical lens for illumination. Further development and improvement of the SPIM started in 2004.[5] After this publication by Huisken et al. the technique found wide application and is still adapted to new measurement situations today (see above). Since 2010 a first ultramicroscope with fluorescence excitation and limited resolution[36] and since 2012 a first SPIM are available commercially.[37] A good overview about the development of SPIM is given in ref.[38] During 2012 also open source projects have started to appear that freely publish complete construction plans for LSFMs and also the required software suites.[39][40][41][42]
Applications
SPIM/LSFM is often used in developmental biology, where it enables long-time (several days) observations of embryonic development (even with full lineage tree reconstruction).[5][43] SPIM can also be combined with techniques, like fluorescence correlation spectroscopy to allow spatially resolved mobility measurements of fluorescing particles (e.g. fluorescent beads, quantum dots or fluorescent proteins) inside living biological samples.[19][20]
Strongly scattering biological tissue such as brain or kidney has to be chemically fixed and cleared before it can be imaged in a SPIM.[44] Special tissue clearing techniques have been developed for this purpose, e.g. 3DISCO, CUBIC and CLARITY. Depending on the index of refraction of the cleared sample, matching immersion fluids and special long-distance objectives must be used during imaging.
 A mouse brain (Thy-1 GFP-M) cleared using 3DISCO method and imaged by light-sheet microscopy.
A mouse brain (Thy-1 GFP-M) cleared using 3DISCO method and imaged by light-sheet microscopy.
References
- Fadero, T.C.; et al. (2018). "LITE microscopy: Tilted light-sheet excitation of model organisms offers high resolution and low photobleaching". The Journal of Cell Biology. 217 (5): 1869–1882. doi:10.1083/jcb.201710087. PMC 5940309. PMID 29490939.
- Keller, Philipp J.; Stelzer, Ernst H. K. (2006). "Lichtscheiben-Mikroskopie in der molekularen Zellbiophysik" (PDF). Laborwelt. 7 (5): 18–21.
- Tomer, Raju; Lovett-Barron, Matthew; Kauvar, Isaac; Andalman, Aaron; Burns, Vanessa M.; Sankaran, Sethuraman; Grosenick, Logan; Broxton, Michael; Yang, Samuel; Deisseroth, Karl (2015). "SPED Light Sheet Microscopy: Fast Mapping of Biological System Structure and Function". Cell. 163 (7): 1796–1806. doi:10.1016/j.cell.2015.11.061. ISSN 0092-8674. PMC 4775738. PMID 26687363.
- A. H. Voie; D. H. Burns; F. A. Spelman (June 1993). "Orthogonal-plane fluorescence optical sectioning: Three-dimensional imaging of macroscopic biological specimens". Journal of Microscopy. 170 (3): 229–236. doi:10.1111/j.1365-2818.1993.tb03346.x. ISSN 0022-2720. PMID 8371260. S2CID 2901024.
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E. H. (2004). "Optical sectioning deep inside live embryos by selective plane illumination microscopy". Science. 305 (5686): 1007–1009. Bibcode:2004Sci...305.1007H. CiteSeerX 10.1.1.456.2250. doi:10.1126/science.1100035. PMID 15310904. S2CID 3213175.
- Timo Mappes; Norbert Jahr; Andrea Csaki; Nadine Vogler; Juergen Popp; Wolfgang Fritzsche (5 November 2012). "The Invention of Immersion Ultramicroscopy in 1912-The Birth of Nanotechnology?". Angewandte Chemie International Edition. 51 (45): 11208–11212. doi:10.1002/anie.201204688. ISSN 1433-7851. PMID 23065955.
- Greger, K; Swoger, J; Stelzer, EH (February 2007). "Basic building units and properties of a fluorescence single plane illumination microscope". Rev Sci Instrum. 78 (2): 023705–023705–7. Bibcode:2007RScI...78b3705G. doi:10.1063/1.2428277. PMID 17578115.
- Huisken, Jan; Stainier, Didier Y. R. (2007). "Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM)". Optics Letters. 32 (17): 2608–10. Bibcode:2007OptL...32.2608H. doi:10.1364/OL.32.002608. ISSN 0146-9592. PMID 17767321. S2CID 15231468.(subscription required)
- Tomer, Raju; Khairy, Khaled; Amat, Fernando; Keller, Philipp J (3 June 2012). "Quantitative high-speed imaging of entire developing embryos with simultaneous multiview light-sheet microscopy". Nature Methods. 9 (7): 755–763. doi:10.1038/nmeth.2062. ISSN 1548-7091. PMID 22660741. S2CID 14191130.(subscription required)
- Krzic, Uros; Gunther, Stefan; Saunders, Timothy E; Streichan, Sebastian J; Hufnagel, Lars (3 June 2012). "Multiview light-sheet microscope for rapid in toto imaging". Nature Methods. 9 (7): 730–733. doi:10.1038/nmeth.2064. ISSN 1548-7091. PMID 22660739. S2CID 13388657.(subscription required)
- Keller, P. J.; Schmidt, A. D.; Wittbrodt, J.; Stelzer, E.H.K. (14 November 2008). "Reconstruction of Zebrafish Early Embryonic Development by Scanned Light Sheet Microscopy" (PDF). Science. 322 (5904): 1065–1069. Bibcode:2008Sci...322.1065K. doi:10.1126/science.1162493. ISSN 0036-8075. PMID 18845710. S2CID 7594561.
- Fahrbach, F. O.; Rohrbach, A. (November 2010). "A line scanned light-sheet microscope with phase shaped self-reconstructing beams". Optics Express. 18 (23): 24229–24244. Bibcode:2010OExpr..1824229F. doi:10.1364/oe.18.024229. PMID 21164769.
- Planchon, T. A.; Gao, L.; Milkie, D. E.; Davidson, M. W.; Galbraith, J. A.; Galbraith, C. G.; Betzig, E. (2011). "Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination". Nature Methods. 8 (5): 417–423. doi:10.1038/nmeth.1586. PMC 3626440. PMID 21378978.
- Vettenburg, Tom; Dalgarno, Heather I C; Nylk, Jonathan; Coll-Lladó, Clara; Ferrier, David E K; Čižmár, Tomáš; Gunn-Moore, Frank J; Dholakia, Kishan (2014). "Light-sheet microscopy using an Airy beam" (PDF). Nature Methods. 11 (5): 541–544. doi:10.1038/nmeth.2922. hdl:10023/5521. PMID 24705473. S2CID 205422713.
- Nylk, Jonathan; McCluskey, Kaley; Preciado, Miguel A.; Mazilu, Michael; Yang, Zhengyi; Gunn-Moore, Frank J.; Aggarwal, Sanya; Tello, Javier A.; Ferrier, David E. K. (1 April 2018). "Light-sheet microscopy with attenuation-compensated propagation-invariant beams". Science Advances. 4 (4): eaar4817. arXiv:1708.02612. Bibcode:2018SciA....4.4817N. doi:10.1126/sciadv.aar4817. PMC 5938225. PMID 29740614.
- Dunsby, C. (2008). "Optically sectioned imaging by oblique plane microscopy". Optics Express. 16 (25): 20306–16. Bibcode:2008OExpr..1620306D. doi:10.1364/OE.16.020306. hdl:10044/1/53595. ISSN 1094-4087. PMID 19065169.
- Zeno Lavagnino; Francesca Cella Zanacchi; Emiliano Ronzitti; Alberto Diaspro (2013). "Two-photon excitation selective plane illumination microscopy (2PE-SPIM) of highly scattering samples: characterization and application". Optics Express. 21 (5): 5998–6008. Bibcode:2013OExpr..21.5998L. doi:10.1364/OE.21.005998. ISSN 1094-4087. PMID 23482168.
- Wolf S, Supatto W, Debregeas G, Mahou P, Kruglik SG, Sintes J, Beaurepaire E, Candelier R (May 2015). "Whole-brain functional imaging with two-photon light-sheet microscopy". Correspondence. Nature Methods. 12 (5): 379–80. doi:10.1038/nmeth.3371. PMID 25924070. S2CID 19746295.
- Capoulade, J.; Wachsmuth, M.; Hufnagel, L.; Knop, M. (2011). "Quantitative fluorescence imaging of protein diffusion and interaction in living cells". Nature Biotechnology. 29 (9): 835–839. doi:10.1038/nbt.1928. PMID 21822256. S2CID 10493584.
- Wohland, T.; Shi, X.; Sankaran, J.; Stelzer, E. H. (May 2010). "Single plane illumination fluorescence correlation spectroscopy (SPIM-FCS) probes inhomogeneous three-dimensional environments". Optics Express. 18 (10): 10627–10641. Bibcode:2010OExpr..1810627W. doi:10.1364/oe.18.010627. PMID 20588915.
- Klaus Greger; Manuel J. Neetz; Emmanuel G. Reynaud; Ernst H.K. Stelzer (2011). "Three-dimensional Fluorescence Lifetime Imaging with a Single Plane Illumination Microscope provides an improved Signal to Noise Ratio". Optics Express. 19 (21): 20743–50. Bibcode:2011OExpr..1920743G. doi:10.1364/OE.19.020743. ISSN 1094-4087. PMID 21997084.
- Francesca Cella Zanacchi; Zeno Lavagnino; Michela Perrone Donnorso; Alessio Del Bue; Laura Furia; Mario Faretta; Alberto Diaspro (9 October 2011). "Live-cell 3D super-resolution imaging in thick biological samples". Nature Methods. 8 (12): 1047–1049. doi:10.1038/nmeth.1744. ISSN 1548-7091. PMID 21983925. S2CID 205420075.
- Jerome Mertz; Jinhyun Kim (2010). "Scanning light-sheet microscopy in the whole mouse brain with HiLo background rejection". Journal of Biomedical Optics. 15 (1): 016027–016027–7. Bibcode:2010JBO....15a6027M. doi:10.1117/1.3324890. ISSN 1083-3668. PMC 2917465. PMID 20210471.
- Friedrich, Mike; Gan, Qiang; Ermolayev, Vladimir; Harms, Gregory S. (2011). "STED-SPIM: Stimulated Emission Depletion Improves Sheet Illumination Microscopy Resolution". Biophysical Journal. 100 (8): L43–5. Bibcode:2011BpJ...100L..43F. doi:10.1016/j.bpj.2010.12.3748. PMC 3077687. PMID 21504720..
- Fadero TC, et al. 2017. "LITE microscopy: a technique for high numerical aperture, low photobleaching fluorescence imaging" bioRxiv doi: https://doi.org/10.1101/181644 https://www.biorxiv.org/content/early/2017/10/04/181644.1
- Jorand R, et al. 2012. "Deep and Clear Optical Imaging of Thick Inhomogeneous Samples" PLOS one doi:https://doi.org/10.1371/journal.pone.0035795
- Jorand R, 2013. "Amélioration des voies de détection et d'illumination d'un microscope SPIM pour l'imagerie 3D des sphéroïdes" theses.fr https://www.theses.fr/180545140
- Alexis Maizel, Daniel von Wangenheim, Fern n Federici, Jim Haseloff, Ernst H.K. Stelzer (October 2011). "High-resolution live imaging of plant growth in near physiological bright conditions using light sheet fluorescence microscopy". The Plant Journal. 68 (2): 377–385. doi:10.1111/j.1365-313X.2011.04692.x. ISSN 0960-7412. PMID 21711399.CS1 maint: multiple names: authors list (link)
- Terrence F. Holekamp; Diwakar Turaga; Timothy E. Holy (13 March 2008). "Fast Three-Dimensional Fluorescence Imaging of Activity in Neural Populations by Objective-Coupled Planar Illumination Microscopy". Neuron. 57 (5): 661–672. doi:10.1016/j.neuron.2008.01.011. ISSN 0896-6273. PMID 18341987. S2CID 9571663.
- Y. Wu; A. Ghitani; R. Christensen; A. Santella; Z. Du; G. Rondeau; Z. Bao; D. Colon-Ramos; H. Shroff (25 October 2011). "Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans". Proceedings of the National Academy of Sciences. 108 (43): 17708–17713. Bibcode:2011PNAS..10817708W. doi:10.1073/pnas.1108494108. ISSN 0027-8424. PMC 3203761. PMID 22006307.
- Wu, Yicong; Wawrzusin, Peter; Senseney, Justin; Fischer, Robert S; Christensen, Ryan; Santella, Anthony; York, Andrew G; Winter, Peter W; Waterman, Clare M; Bao, Zhirong; Colón-Ramos, Daniel A; McAuliffe, Matthew; Shroff, Hari (2013). "Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy". Nature Biotechnology. 31 (11): 1032–1038. doi:10.1038/nbt.2713. ISSN 1087-0156. PMC 4105320. PMID 24108093.
- "Webpage Huisken Lab". Archived from the original on 7 July 2014.
- Jörg G. Ritter; Roman Veith; Jan-Peter Siebrasse; Ulrich Kubitscheck (2008), "High-contrast single-particle tracking by selective focal plane illumination microscopy", Optics Express, 16 (10), pp. 7142–52, Bibcode:2008OExpr..16.7142R, doi:10.1364/OE.16.007142, ISSN 1094-4087, PMID 18545417
- Jerome Fehrenbach; Pierre Weiss; Corinne Lorenzo (2012). "Variational Algorithms to Remove Stationary Noise: Applications to Microscopy Imaging" (PDF). IEEE Transactions on Image Processing. 21 (10): 4420–4430. Bibcode:2012ITIP...21.4420F. doi:10.1109/TIP.2012.2206037. ISSN 1057-7149. PMID 22752131. S2CID 6828193.
- Nobel lecture of R. A. Zsigmondy: Properties of colloids (including a short explanation of the ultramicroscope)
- press release of LaVision Biotech Archived 24 December 2013 at the Wayback Machine (accessed 2012-11-04)
- Carl Zeiss press release about the Lightsheet Z.1 Light Sheet Microscope System (accessed 2012-11-15)
- P. A. Santi (1 February 2011). "Light Sheet Fluorescence Microscopy: A Review". Journal of Histochemistry & Cytochemistry. 59 (2): 129–138. doi:10.1369/0022155410394857. ISSN 0022-1554. PMC 3201139. PMID 21339178.
- OpenSPIM project webpage (accessed 2013-06-08)
- Peter G Pitrone; Johannes Schindelin; Luke Stuyvenberg; Stephan Preibisch; Michael Weber; Kevin W Eliceiri; Jan Huisken; Pavel Tomancak (9 June 2013). "OpenSPIM: an open-access light-sheet microscopy platform". Nature Methods. 10 (7): 598–9. arXiv:1302.1987. doi:10.1038/nmeth.2507. ISSN 1548-7091. PMC 7450513. PMID 23749304.
- The OpenSPIN project webpage (accessed 2013-06-08)
- Emilio J Gualda, Tiago Vale, Pedro Almada, Jos A Feij, Gabriel G Martins, Nuno Moreno (9 June 2013). "OpenSpinMicroscopy: an open-source integrated microscopy platform". Nature Methods. 10 (7): 599–600. doi:10.1038/nmeth.2508. ISSN 1548-7091. PMID 23749300. S2CID 27935584.CS1 maint: multiple names: authors list (link)
- Verveer, P. J.; Swoger, J.; Pampaloni, F.; Greger, K.; Marcello, M.; Stelzer, E. H. (2007). "High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy". Nature Methods. 4 (4): 311–313. doi:10.1038/nmeth1017. PMID 17339847. S2CID 12440781.
- Jährling, Nina; Becker, Klaus; Wegenast-Braun, Bettina M.; Grathwohl, Stefan A.; Jucker, Mathias; Dodt, Hans-Ulrich (27 May 2015). Vitorica, Javier (ed.). "Cerebral β-Amyloidosis in Mice Investigated by Ultramicroscopy". PLOS ONE. 10 (5): e0125418. Bibcode:2015PLoSO..1025418J. doi:10.1371/journal.pone.0125418. ISSN 1932-6203. PMC 4446269. PMID 26017149.
- Corinne Lorenzo; Céline Frongia; Raphael Jorand; Jérome Fehrenbach; Pierre Weiss; Amina Maandhui; Guillaume Gay; Bernard Ducommun; Valérie Lobjois (2011). "Live cell division dynamics monitoring in 3D large spheroid tumor models using light sheet microscopy". Cell Division. 6 (1): 22. doi:10.1186/1747-1028-6-22. ISSN 1747-1028. PMC 3274476. PMID 22152157.
- Daisuke Takao; Atsushi Taniguchi; Takaaki Takeda; Seiji Sonobe; Shigenori Nonaka; Alexandre J. Kabla (5 December 2012), "High-Speed Imaging of Amoeboid Movements Using Light-Sheet Microscopy", PLOS ONE, 7 (12), p. e50846, Bibcode:2012PLoSO...750846T, doi:10.1371/journal.pone.0050846, ISSN 1932-6203, PMC 3515486, PMID 23227214
Further reading
- Review: P. A. Santi (1 February 2011). "Light Sheet Fluorescence Microscopy: A Review". Journal of Histochemistry & Cytochemistry. 59 (2): 129–138. doi:10.1369/0022155410394857. ISSN 0022-1554. PMC 3201139. PMID 21339178.
- Review of different LSFM modalities and results in developmental biology: Huisken, J.; Stainier, D.Y.R. (22 May 2009). "Selective plane illumination microscopy techniques in developmental biology". Development. 136 (12): 1963–1975. doi:10.1242/dev.022426. ISSN 0950-1991. PMC 2685720. PMID 19465594.
- Review of LSFM for imaging anatomic structures: Buytaert, J.; Descamps, Emilie; Adriaens, Dominique; Dirckx, Joris J. J. (12 August 2011). "The OPFOS Microscopy Family: High-Resolution Optical Sectioning of Biomedical Specimens". Anatomy Research International. 2012: 206238–(1–9). arXiv:1106.3162. Bibcode:2011arXiv1106.3162B. doi:10.1155/2012/206238. PMC 3335623. PMID 22567307.
- Editorial: "Method of the Year 2014". Nature Methods. 12 (1): 1. 30 December 2014. doi:10.1038/nmeth.3251. PMID 25699311.

External links
- Video of a typical experiment in developmental biology, using a SPIM on YouTube: The linked video shows the development of a fruit fly embryo, which was recorded during 20 hours. Two projections of the full 3D dataset are shown.
- The mesoSPIM Initiative. Open-source light-sheet microscopes for imaging cleared tissue.
Optical microscopy | ||
|---|---|---|
| Illumination and contrast methods |  | |
| Fluorescence methods | ||
| Sub-diffraction limit techniques | ||