Lymphangiomatosis
Lymphangiomatosis is a condition where a lymphangioma is not present in a single localised mass, but in a widespread or multifocal manner. It is a rare type of tumor which results from an abnormal development of the lymphatic system.[1]
| Lymphangiomatosis | |
|---|---|
| Other names | LYMF |
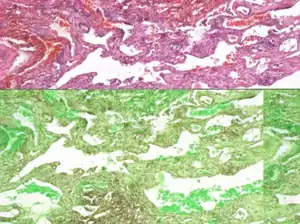 | |
| Lung biopsy showing infiltration of lymphatic tissue. | |
It is thought to be the result of congenital errors of lymphatic development occurring prior to the 20th week of gestation.[2] Lymphangiomatosis is a condition marked by the presence of cysts that result from an increase both in the size and number of thin-walled lymphatic channels that are abnormally interconnected and dilated.[2][3][4] 75% of cases involve multiple organs.[2] It typically presents by age 20 and, although it is technically benign, these deranged lymphatics tend to invade surrounding tissues and cause problems due to invasion and/or compression of adjacent structures.[2] The condition is most common in the bones and lungs[2] and shares some characteristics with Gorham’s disease. Up to 75% of patients with lymphangiomatosis have bone involvement, leading some to conclude that lymphangiomatosis and Gorham’s disease should be considered as a spectrum of disease rather than separate diseases.[2][5] When it occurs in the lungs, lymphangiomatosis has serious consequences and is most aggressive in the youngest children.[2][4] When the condition extends into the chest it commonly results in the accumulation of chyle in the linings of the heart and/or lungs.[2][4]
Chyle is composed of lymph fluid and fats that are absorbed from the small intestine by specialized lymphatic vessels called lacteals during digestion. The accumulations are described based on location: chylothorax is chyle in the chest; chylopericardium is chyle trapped inside the sack surrounding the heart; chyloascites is chyle trapped in the linings of the abdomen and abdominal organs. The presence of chyle in these places accounts for many of the symptoms and complications associated with both lymphangiomatosis and Gorham’s disease.[2][6] The incidence of lymphangiomatosis is unknown and it is often misdiagnosed. It is separate and distinct from lymphangiectasis, lymphangioleiomyomatosis (LAM), pulmonary capillary hemangiomatosis, Kaposi’s sarcoma, and kaposiform hemangioendothelioma.[4] Its unusual nature makes lymphangiomatosis (and Gorham’s disease) a diagnostic and therapeutic challenge.[4][7] A multidisciplinary approach is generally necessary for optimal diagnosis and symptom management. The term literally means lymphatic system (lymph) vessel (angi) tumor or cyst (oma) condition (tosis).
Signs and symptoms
Lymphangiomatosis is a multi-system disorder. Symptoms depend on the organ system involved and, to varying degrees, the extent of the disease. Early in the course of the disease patients are usually asymptomatic, but over time the abnormally proliferating lymphatic channels that constitute lymphangiomatosis are capable of massive expansion and infiltration into surrounding tissues, bone, and organs.[2] Because of its slow course and often vague symptoms, the condition is frequently under-recognized or misdiagnosed.[8]
Early signs of disease in the chest include wheezing, cough, and feeling short of breath, which is often misdiagnosed as asthma.[2] The pain that accompanies bone involvement may be attributed to "growing pains" in younger children. With bone involvement the first indication for disease may be a pathological fracture. Symptoms may not raise concern, or even be noted, until the disease process has advanced to a point where it causes restrictive compression of vital structures. Further, the occurrence of chylous effusions seems to be unrelated to the pathologic "burden" of the disease, the extent of involvement in any particular tissue or organ, or the age of the patient.[9] This offers one explanation as to why, unfortunately, the appearance of chylous effusions in the chest or abdomen may be the first evidence of the disease.
Following are some of the commonly reported symptoms of lymphangiomatosis, divided into the regions/systems in which the disease occurs:
Heart and chest
Symptoms that arise from disease of the cardiothoracic region include a chronic cough, wheezing, dyspnea (shortness of breath)—especially serious when occurring at rest or when lying down—fever, chest pain, rapid heartbeat, dizziness, anxiety, and coughing up blood or chyle.[2] As the deranged lymphatic vessels invade the organs and tissues in the chest they put stress on the heart and lungs, interfering with their ability to function normally. Additionally, these lymphatic vessels may leak, allowing fluid to accumulate in the chest, which puts further pressure on the vital organs, thus increasing their inability to function properly.[2] Accumulations of fluid and chyle are named based on their contents and location: pulmonary edema (the presence of fluid and/or chyle in the lung), pleural effusions (fluid in the lung lining), pericardial effusions (fluid in the heart sack), chylothorax (chyle in the pleural cavity); and chylopericardium (chyle in the heart sack).
Abdominal
Lymphangiomatosis has been reported in every region of the abdomen, though the most reported sites involve the intestines and peritoneum; spleen, kidneys, and liver. Often there are no symptoms until late in the progression of the disease. When they do occur, symptoms include abdominal pain and/or distension; nausea, vomiting, diarrhea; decreased appetite and malnourishment. When the disease affects the kidneys the symptoms include flank pain, abdominal distension, blood in the urine, and, possibly, elevated blood pressure, which may result in it being confused with other cystic renal disease.[10] When lymphangiomatosis occurs in the liver and/or spleen it may be confused with polycystic liver disease.[11] Symptoms may include abdominal fullness and distension; anemia, disseminated intravascular coagulopathy (DIC), fluid accumulation in the abdomen(ascites), decreased appetite, weight loss, fatigue; late findings include liver failure.[2][11][12]
Bones
Symptoms of lymphangiomatosis in the skeletal system are the same as those of Gorham’s disease. Frequently asymptomatic, skeletal lymphangiomatosis may be discovered incidentally or when a pathological fracture occurs. Patients may experience pain of varying severity in areas around the effected bone. When the disease occurs in the bones of the spine, neurological symptoms such as numbness and tingling may occur due to spinal nerve compression.[13] Progression of disease in the spine may lead to paralysis. Lymphangiomatosis in conjunction with Chiari I malformation also has been reported.[14]
Causes
The cause of lymphangiomatosis is not yet known. As stated earlier, it is generally considered to be the result of congenital errors of lymphatic development occurring prior to the 20th week of gestation.[2] However, the root causes of these conditions remains unknown and further research is necessary.
Diagnosis
Because it is rare and has a wide spectrum of clinical, histological, and imaging features, diagnosing lymphangiomatosis can be challenging.[15] Plain x-rays reveal the presence of lytic lesions in bones, pathological fractures, interstitial infiltrates in the lungs, and chylous effusions that may be present even when there are no outward symptoms.[2][5][7]
The most common locations of lymphangiomatosis are the lungs and bones and one important diagnostic clue is the coexistence of lytic bone lesions and chylous effusion.[2] An isolated presentation usually carries a better prognosis than does multi-organ involvement; the combination of pleural and peritoneal involvement with chylous effusions and lytic bone lesions carries the least favorable prognosis.[16]
When lung involvement is suspected, high resolution computed tomography (HRCT) scans may reveal a diffuse liquid-like infiltration in the mediastinal and hilar soft tissue, resulting from diffuse proliferation of lymphatic channels and accumulation of lymphatic fluid; diffuse peribronchovascular and interlobular septal thickening; ground-glass opacities; and pleural effusion.[2][17] Pulmonary function testing reveals either restrictive pattern or a mixed obstructive/restrictive pattern.[2][4] While x-rays, HRCT scan, MRI, ultrasound, lymphangiography, bone scan, and bronchoscopy all can have a role in identifying lymphangiomatosis, biopsy remains the definitive diagnostic tool.[2][5][7][17][18][19]
Microscopic examination of biopsy specimens reveals an increase in both the size and number of thin walled lymphatic channels along with lymphatic spaces that are interconnecting and dilated, lined by a single attenuated layer of endothelial cells involving the dermis, subcutis, and possibly underlying fascia and skeletal muscle.[3] Additionally, Tazelaar, et al., described a pattern of histological features of lung specimens from nine patients in whom no extrathoracic lesions were identified, which they termed "diffuse pulmonary lymphangiomatosis" (DPL).[4]
Recognition of the disease requires a high index of suspicion and an extensive workup. Because of its serious morbidity, lymphangiomatosis must always be considered in the differential diagnosis of lytic bone lesions accompanied by chylous effusions, in cases of primary chylopericardium, and as part of the differential diagnosis in pediatric patients presenting with signs of interstitial lung disease.[2][18][20][21]
Treatment
There is no standard approach to the treatment of lymphangiomatosis and treatment often is aimed at reducing symptoms.[2][17] Surgical intervention may be indicated when complications arise and a number of reports of response to surgical interventions, medications, and dietary approaches can be found in the medical literature.[2][16][17]
Unfortunately, there is no standardized treatment for lymphangiomatosis and no cure. Treatment modalities that have been reported in the medical literature, by system, include:
Heart and chest
Thoracocentesis, pericardiocentesis, pleurodesis, ligation of thoracic duct, pleuroperitoneal shunt, radiation therapy, pleurectomy, pericardial window, pericardiectomy, thalidomide, interferon alpha 2b, Total Parenteral Nutrition (TPN), medium chain triglyceride (MCT) and high protein diet, chemotherapy, sclerotherapy, transplant;
Abdominal
interferon alpha 2b, sclerotherapy, resection, percutaneous drainage, Denver shunt, Total Parenteral Nutrition (TPN), medium chain triglyceride (MCT) and high protein diet, transplant, splenectomy;
Bone
interferon alpha 2b, bisphosphonates (i.e. pamidronate), surgical resection, radiation therapy, sclerotherapy, percutaneous bone cement, bone grafts, prosthesis, surgical stabilization.
Epidemiology
Lymphangiomatosis can occur at any age, but the incidence is highest in children and teenagers. Signs and symptoms are typically present before the age of 20 and the condition is often under-recognized in adults.[2]
It affects males and females of all races and exhibits no inheritance pattern. The medical literature contains case reports from every continent.
Because it is so rare, and commonly misdiagnosed, it is not known exactly how many people are affected by this disease.
References
- Marom, EM; Moran, CA; Munden, RF (April 2004). "Generalized lymphangiomatosis". American Journal of Roentgenology. 182 (4): 1068. doi:10.2214/ajr.182.4.1821068. PMID 15039189.
- Faul J.L., Berry G.J., Colby T.V., Ruoss S.J., Walter M.B, Rosen G.D., Raffin T.A. (2000). "Thoracic Lymphangiomas, Lymphangiectasis, Lymphangiomatosis, and Lymphatic Dysplasia Syndrome". Am. J. Respir. Crit. Care Med. 161 (3): 1037–1046. doi:10.1164/ajrccm.161.3.9904056. PMID 10712360.CS1 maint: multiple names: authors list (link)
- Pernick, Nat. "Soft Tissue Tumors Part 3 Muscle, Vascular, Nerve, Other Lymphangiomatosis." PathologyOutlines.com. PathologyOutlines.com, Inc., 10/17/2009. Web. 6 Sep 2011. http://www.pathologyoutlines.com/topic/softtissue3lymphangiomatosis.html.
- Tazelaar HD, Kerr D, Yousem SA, Saldana MJ, Langston C, Colby TV (Dec 1993). "Diffuse pulmonary lymphangiomatosis". Hum Pathol. 24 (12): 1313–22. doi:10.1016/0046-8177(93)90265-i. PMID 8276379.CS1 maint: multiple names: authors list (link)
- Aviv RI, McHugh K, Hunt J. Angiomatosis of bone and soft tissue: a spectrum of disease from diffuse lymphangiomatosis to vanishing bone disease in young patients. Clin Radiol. 2001 Mar;56(3):184-90.
- Duffy B, Manon R, Patel R, Welsh JS, et al. A case of Gorham’s disease with chylothorax treated curatively with radiation therapy. Clin Med Res. 2005;3:83–
- Yeager ND, Hammond S, Mahan J, Davis JT, Adler B. Unique diagnostic features and successful management of a patient with disseminated lymphangiomatosis and chylothorax. J Pediatr Hematol Oncol. 2008 Jan;30(1):66-9.
- Venkatramani R, Ma NS, Pitukcheewanont P, Malogolowkin MH, Mascarenhas L. Gorham’s disease and diffuse lymphangiomatosis in children and adolescents. Pediatr Blood Cancer. 2011 Apr;56(4):667-70
- Zisis C, Spiliotopoulos K, Patronis M, Filippakis G, Stratakos G, Tzelepis G, Bellenis I. Diffuse lymphangiomatosis: are there any clinical or therapeutic standards? J Thorac Cardiovasc Surg. 2007 Jun;133(6):1664-5.
- Hakeem A, Gojwari TA, Reyaz S, Rasool S, Shafi H, Mufti S (Jan 2010). "Computed tomography findings in bilateral perinephric lymphangiomatosis". Urol. Ann. 2 (1): 26–8. doi:10.4103/0974-7796.62922. PMC 2934585. PMID 20842254.CS1 maint: multiple names: authors list (link)
- O’Sullivan DA, Torres VE, de Groen PC, Batts KP, King BF, Vockley J. "Hepatic lymphangiomatosis mimicking polycystic liver disease. Mayo Clin Proc. 1998 Dec;73(12):1188-92.
- Miller C, Mazzaferro V, Makowka L, ChapChap P, Demetris J, Tzakis A, Esquivel CO, Iwatsuki S, Starzl TE (Apr 1988). "Orthotopic liver transplantation for massive hepatic lymphangiomatosis". Surgery. 103 (4): 490–5. PMC 2963582. PMID 3281302.CS1 maint: multiple names: authors list (link)
- Watkins RG 4th, Reynolds RA, McComb JG, Tolo VT. Lymphangiomatosis of the spine: two cases requiring surgical intervention. Spine. 2003 Feb 1;28(3):E45-50.
- Jea A, McNeil A, Bhatia S, Birchansky S, Sotrel A, Ragheb J, Morrison G. A rare case of lymphangiomatosis of the craniocervical spine in conjunction with a Chiari I malformation. Pediatr Neurosurg. 2003 Oct;39(4):212-5.
- Shah V, Shah S, Barnacle A, Sebire NJ, Brock P, Harper JI, McHugh K. Mediastinal involvement in lymphangiomatosis: a previously unreported MRI sign. Pediatr Radiol. 2011 Aug;41(8):985-92.
- CS Wong, TYC Chu. Clinical and radiological features of generalised lymphangiomatosis. Hong Kong Med J 2008;14:402-4
- Ming-hua DU, Ruan-jian YE, Kun-kun SUN, Jian-feng LI, Dan-hua Shen, Jun Wang, Zhan-cheng GAO (2011). "Diffuse pulmonary lymphangiomatosis: a case report with literature review". Chinese Medical Journal. 124 (5): 797–800. PMID 21518582.CS1 maint: multiple names: authors list (link)
- Swensen SJ, Hartman TE, Mayo JR, Colby TV, Tazelaar HD, Müller NL (1995). "Diffuse pulmonary lymphangiomatosis: CT findings". J Comput Assist Tomogr. 19 (3): 348–52. doi:10.1097/00004728-199505000-00002. PMID 7790540. S2CID 13399987.CS1 maint: multiple names: authors list (link)
- Wunderbaldinger P, Paya K, Partik B, Turetschek K, Hörmann M, Horcher E, Bankier AA (Mar 2000). "CT and MR imaging of generalized cystic lymphangiomatosis in pediatric patients". Am J Roentgenol. 174 (3): 827–32. doi:10.2214/ajr.174.3.1740827. PMID 10701634.CS1 maint: multiple names: authors list (link)
- Martinez-Pajares JD, Rosa-Camacho V, Camacho-Alonso JM, Zabala-Arguelles I, Gil-Jaurena JM, Milano-Manso G. Spontaneous chylous pericardial effusion: report of two cases. An Pediatr (Barc). 2010 Jul;73(1):42-6.
- Lynch DA, Hay T, Newell JD (Sep 1999). "Jr, Divgi VD, Fan LL. Pediatric diffuse lung disease: diagnosis and classification using high-resolution CT". Am J Roentgenol. 173 (3): 713–8. doi:10.2214/ajr.173.3.10470910. PMID 10470910.CS1 maint: multiple names: authors list (link)
Further reading
- Dellinger M. T., Garg N, Olsen B.R. (2014). "Viewpoints on vessels and vanishing bones in Gorham-Stout disease". Bone. 63C: 47–52. doi:10.1016/j.bone.2014.02.011. PMID 24583233.CS1 maint: multiple names: authors list (link)
- Trenor C, Chaudry G (Aug 2014). "Complex lymphatic anomalies". Semin Pediatr Surg. 23 (4): 186–90. doi:10.1053/j.sempedsurg.2014.07.006. PMID 25241096.
- Lala S, Mulliken JB, Alomari AI, Fishman SJ, Kozakewich HP, Chaudry G (Jul 2013). "Gorham-Stout disease and generalized lymphatic anomaly—clinical, radiologic, and histologic differentiation". Skeletal Radiol. 42 (7): 917–24. doi:10.1007/s00256-012-1565-4. PMID 23371338. S2CID 22640055.CS1 maint: multiple names: authors list (link)
External Resources
- Lymphangiomatosis & Gorham's Disease Alliance (LGDA) is a 501(c)(3) organization incorporated in the United States and serving patients and families around the world: www.lgdalliance.org
- International LGDA Registry for Lymphatic Malformations is a patient outreach and research development project of the LGDA: www.lgdaregistry.org