Müllerian agenesis
Müllerian agenesis, also known as Mayer–Rokitansky–Küster–Hauser syndrome (MRKH) or vaginal agenesis, is a congenital malformation characterized by a failure of the Müllerian duct to develop, resulting in a missing uterus and variable degrees of vaginal hypoplasia of its upper portion. Müllerian agenesis (including absence of the uterus, cervix and/or vagina) is the cause in 15% of cases of primary amenorrhoea.[2] Because most of the vagina does not develop from the Müllerian duct, instead developing from the urogenital sinus, along with the bladder and urethra, it is present even when the Müllerian duct is completely absent. Because ovaries do not develop from the Müllerian ducts, affected people might have normal secondary sexual characteristics but are infertile due to the lack of a functional uterus. However, parenthood is possible through use of gestational surrogates.
| Müllerian agenesis | |
|---|---|
| Other names | Mayer–Rokitansky–Küster–Hauser syndrome (MRKH), Rokitansky–Küster–Hauser syndrome (RKH or RKHS), müllerian aplasia, vaginal agenesis |
| Specialty | Gynecology |
| Frequency | 1 in 4,500 females[1] |
Mayer–Rokitansky–Küster–Hauser syndrome is hypothesized to be a result of autosomal dominant inheritance with incomplete penetrance and variable expressivity, which contributes to the complexity involved in identifying of the underlying mechanisms causing the condition. Because of the variance in inheritance, penetrance and expressivity patterns, MRKH is subdivided into two types: type 1, in which only the structures developing from the Müllerian duct are affected (the upper vagina, cervix, and uterus), and type 2, where the same structures are affected, but is characterized by the additional malformations of other body systems most often including the renal and skeletal systems. MRKH type 2 includes MURCS (Müllerian Renal Cervical Somite).
The majority of MRKH syndrome cases are characterized as sporadic, but familial cases have provided evidence that, at least for some patients, MRKH is an inherited disorder. The underlying causes of MRKH syndrome is still being investigated, but several causative genes have been studied for their possible association with the syndrome. Most of these studies have served to rule-out genes as causative factors in MRKH, but thus far, only WNT4 has been associated with MRKH with hyperandrogenism.[3][4]
Reports of MRKH syndrome can be traced back to Hippocrates (460 B.C.–377 B.C.).[5][6] The medical eponym honors August Franz Josef Karl Mayer (1787–1865), Carl Freiherr von Rokitansky (1804–1878), Hermann Küster (1879–1964) and Georges Andre Hauser (1921–2009).
Signs and symptoms
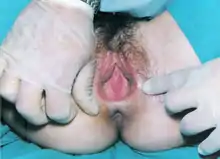
A female with this condition is hormonally normal; that is, the person will enter puberty with development of secondary sexual characteristics including thelarche and pubarche (pubic hair). The person's karyotype will be 46,XX. At least one ovary is intact, if not both, and ovulation usually occurs. Typically, the vagina is shortened and intercourse may, in some cases, be difficult and painful. Medical examination supported by gynecologic ultrasonography demonstrates a complete or partial absence of the cervix, uterus, and vagina.
If there is no uterus, a woman with MRKH cannot carry a pregnancy without intervention. It is possible for the person to have genetic offspring by in vitro fertilization (IVF) and surrogacy. Successful uterine transplant has been performed in limited numbers of patients, resulting in several live births, but the technique is not widespread or accessible to many women.[7]
A person with MRKH typically discovers the condition when, during puberty years, the menstrual cycle does not start (primary amenorrhoea). Some find out earlier through surgeries for other conditions, such as a hernia.
Causes
WNT4 (found on the short arm (p) of chromosome 1) has been clearly implicated in the atypical version of this disorder. A genetic mutation causes a leucine to proline residue substitution at amino acid position 12.[8] This occurrence reduces the intranuclear levels of β catenin. In addition, it removes the inhibition of steroidogenic enzymes like 3β-hydroxysteriod dehydrogenase and 17α-hydroxylase. Patients therefore have androgen excess.[8] Furthermore, without WNT4, the Müllerian duct is either deformed or absent. Female reproductive organs, such as the cervix, fallopian tubes, ovaries, and much of the vagina, are hence affected.[9]
An association with a deletion mutation in the long arm (q) of chromosome 17 (17q12) has been reported. The gene LHX1 is located in this region and may be the cause of a number of these cases.[10]
Diagnosis
Classification
- Typical MRKH – Isolated uterovaginal aplasia/hypoplasia
- Prevalence – 64%
- Atypical MRKH – Uterovaginal aplasia/hypoplasia with renal malformation or uterovaginal aplasia/hypoplasia with ovarian dysfunction
- Prevalence – 24%
- MURCS syndrome – Uterovaginal aplasia/hypoplasia with renal malformation, skeletal malformation, and cardiac malformation
- Prevalence – 12%[8]
Treatment
A number of treatments have become available to create a functioning vagina, but in the absence of a uterus no surgery is currently available to make pregnancy possible. Standard approaches use vaginal dilators and/or surgery to develop a functioning vagina to allow for penetrative sexual intercourse. A number of surgical approaches have been used. In the McIndoe procedure, a skin graft is applied to form an artificial vagina. After the surgery, dilators are still necessary to prevent vaginal stenosis. The Vecchietti procedure has been shown to result in a vagina that is comparable to a normal vagina in patients.[11][12] In the Vecchietti procedure, a small plastic “olive” is threaded against the vaginal area, and the threads are drawn through the vaginal skin, up through the abdomen and through the navel using laparoscopic surgery. There the threads are attached to a traction device. The operation takes about 45 minutes. The traction device is then tightened daily so the olive is pulled inwards and stretches the vagina by approximately 1 cm per day, creating a vagina approximately 7 cm deep in 7 days, although it can be more than this.[13] Another approach is the use of an autotransplant of a resected sigmoid colon using laparoscopic surgery; results are reported to be very good with the transplant becoming a functional vagina.[14]
Uterine transplantation has been performed in a number of people with MRKH, but the surgery is still in the experimental stage.[15] Since ovaries are present, people with this condition can have genetic children through IVF with embryo transfer to a gestational carrier. Some also choose to adopt.[16][17] In October 2014, it was reported that a month earlier a 36-year-old Swedish woman became the first person with a transplanted uterus to give birth to a healthy baby. She was born without a uterus, but had functioning ovaries. She and the father went through IVF to produce 11 embryos, which were then frozen. Doctors at the University of Gothenburg then performed the uterus transplant, the donor being a 61-year-old family friend. One of the frozen embryos was implanted a year after the transplant, and the baby boy was born prematurely at 31 weeks after the mother developed pre-eclampsia.
Promising research include the use of laboratory-grown structures, which are less subject to the complications of non-vaginal tissue, and may be grown using the person's own cells as a culture source.[18][19] The recent development of engineered vaginas using the patient's own cells has resulted in fully functioning vaginas capable of menstruation and orgasm in a number of patients showing promise of fully correcting this condition in some of the sufferers.[20][21]
Epidemiology
The prevalence remains sparsely investigated. To date, two population-based nationwide studies have been conducted both estimating a prevalence about 1 in 5,000 live female births.[22][23] According to some reports, Queen Amalia of Greece may have had the syndrome, but a 2011 review of the historical evidence concludes that it is not possible to determine the inability of her and her husband to have a child.[24][6] Their inability to conceive an heir contributed to the overthrow of the king King Otto.[24]
People with MRKH
- Ben Barres, transgender male scientist[25]
- Esther Morris Leidolf, medical sociologist and intersex activist
- Shon Klose, Australian intersex activist and musician[26][27]
- Jaclyn Schultz, Miss Michigan[28]
Suspected cases
See also
References
- "Müllerian Agenesis: Diagnosis, Management, and Treatment". ACOG. January 2018. Retrieved 21 January 2018.
- Welt CK, Barbieri RL. "Etiology, diagnosis, and treatment of primary amenorrhea". Retrieved 19 November 2015.
- Woten M. "Quick Lesson: Mayer–Rokitansky–Kuster–Hauser Syndrome". In Cinahl DP (ed.). Information Systems.
- Fontana L, Gentilin B, Fedele L, Gervasini C, Miozzo M (February 2017). "Genetics of Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome". Clinical Genetics. 91 (2): 233–246. doi:10.1111/cge.12883. PMID 27716927. S2CID 283394.
- Goldwyn RM (March 1977). "History of attempts to form a vagina". Plastic and Reconstructive Surgery. 59 (3): 319–29. doi:10.1097/00006534-197703000-00002. PMID 320610. S2CID 39727188.
- Patnaik SS, Brazile B, Dandolu V, Ryan PL, Liao J (January 2015). "Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome: a historical perspective". Gene. 555 (1): 33–40. doi:10.1016/j.gene.2014.09.045. PMID 25260227.
- Lewis T. "Uterus transplants: My sister gave me her womb". The Guardian. Retrieved 10 July 2016.
- Sultan C, Biason-Lauber A, Philibert P (January 2009). "Mayer–Rokitansky–Kuster–Hauser syndrome: recent clinical and genetic findings". Gynecological Endocrinology. 25 (1): 8–11. doi:10.1080/09513590802288291. PMID 19165657. S2CID 33461252.
- "WNT4 Müllerian aplasia and ovarian dysfunction". Genetics Home Reference. Retrieved 2012-08-18.
- Ledig S, Brucker S, Barresi G, Schomburg J, Rall K, Wieacker P (2012) Frame shift mutation of LHX1 is associated with Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome. Hum Reprod
- Vecchietti G (1965). "[Creation of an artificial vagina in Rokitansky–Küster–Hauser syndrome]". Attualita di Ostetricia e Ginecologia (in Italian). 11 (2): 131–47. PMID 5319813.
- Fedele L, Bianchi S, Tozzi L, Borruto F, Vignali M (November 1996). "A new laparoscopic procedure for creation of a neovagina in Mayer–Rokitansky–Kuster–Hauser syndrome". Fertility and Sterility. 66 (5): 854–7. doi:10.1016/S0015-0282(16)58653-1. PMID 8893702.
- "Vecchietti Procedure" (PDF). University College University Hospitals. Archived from the original (PDF) on 2009-04-11. Retrieved 2010-04-03..
- Hold MK (2007-01-16). "Modernes Management der angeborenen (Mayer-Rokitansky-Küster-Hauser, MRKH-Syndrom) und erworbenen Vaginalaplasie" (PDF). Frauenheilkunde-Aktuell (in German).
- Ozkan O, Akar ME, Ozkan O, Erdogan O, Hadimioglu N, Yilmaz M, Gunseren F, Cincik M, Pestereli E, Kocak H, Mutlu D, Dinckan A, Gecici O, Bektas G, Suleymanlar G (February 2013). "Preliminary results of the first human uterus transplantation from a multiorgan donor". Fertility and Sterility. 99 (2): 470–6. doi:10.1016/j.fertnstert.2012.09.035. PMID 23084266.
- Akhter N, Begum B (3 February 2013). "Evaluation and management of cases of primary amenorrhoea with MRKH syndrome". Bangladesh Medical Journal Khulna. 45 (1–2): 24–29. doi:10.3329/bmjk.v45i1-2.13626.
- "Rokitansky Syndrome: Information for Parents / Carers" (PDF). Saint Mary's Hospital, Manchester. Archived from the original (PDF) on 19 February 2017. Retrieved 11 April 2014.
- "Laboratory-grown vaginas implanted in patients". Retrieved 14 April 2014.
- Raya-Rivera AM, Esquiliano D, Fierro-Pastrana R, López-Bayghen E, Valencia P, Ordorica-Flores R, Soker S, Yoo JJ, Atala A (July 2014). "Tissue-engineered autologous vaginal organs in patients: a pilot cohort study". Lancet. 384 (9940): 329–36. doi:10.1016/S0140-6736(14)60542-0. PMID 24726478. S2CID 6296110.
- Catherine de Lange (2014). "Engineered vaginas grown in women for the first time". New Scientist.
- Raya-Rivera AM, Esquiliano D, Fierro-Pastrana R, López-Bayghen E, Valencia P, Ordorica-Flores R, Soker S, Yoo JJ, Atala A (July 2014). "Tissue-engineered autologous vaginal organs in patients: a pilot cohort study". Lancet. 384 (9940): 329–36. doi:10.1016/S0140-6736(14)60542-0. PMID 24726478. S2CID 6296110.
- Aittomäki K, Eroila H, Kajanoja P (September 2001). "A population-based study of the incidence of Müllerian aplasia in Finland". Fertility and Sterility. 76 (3): 624–5. doi:10.1016/s0015-0282(01)01963-x. PMID 11570363.
- Herlin M, Bjørn AM, Rasmussen M, Trolle B, Petersen MB (October 2016). "Prevalence and patient characteristics of Mayer–Rokitansky–Küster–Hauser syndrome: a nationwide registry-based study". Human Reproduction. 31 (10): 2384–90. doi:10.1093/humrep/dew220. PMID 27609979.
- Poulakou-Rebelakou E, Tsiamis C, Tompros N, Creatsas G (March 2011). "The lack of a child, the loss of a throne: the infertility of the first royal couple of Greece (1833–62)". The Journal of the Royal College of Physicians of Edinburgh. 41 (1): 73–7. doi:10.4997/JRCPE.2011.115. PMID 21365071.
- Morgan, Jules (2019). "Coming out as a male scientist in a man's world" (PDF). The Lancet. 7 (4): 275–9. doi:10.1016/j.neuron.2013.10.005. PMID 24139033.
- sleath, emma (2014-12-03). "I am intersex: Shon Klose's story". ABC Alice Springs. Retrieved 2019-10-27.
- One Plus One: Shon Klose, Australian Broadcasting Corporation, 2015-09-28, retrieved 2019-10-27
- "Why This Woman Is Proud to Be Known as "The Pageant Queen Without a Uterus"". Cosmopolitan. 2015-01-20. Retrieved 2018-09-09.
- "The infertility of the first royal couple of Greece" (PDF). Royal college of physicians of Edinburgh.
Further reading
- Morcel K, Camborieux L, Guerrier D (March 2007). "Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome". Orphanet Journal of Rare Diseases. 2 (13): 13. doi:10.1186/1750-1172-2-13. PMC 1832178. PMID 17359527.
- Varner RE, Younger JB, Blackwell RE (June 1985). "Müllerian dysgenesis". The Journal of Reproductive Medicine. 30 (6): 443–50. PMID 4020785.
External links
- Online Mendelian Inheritance in Man (OMIM): 277000
- American College of Obstetricians and Gynecologists, Müllerian Agenesis: Resource Overview
- The National Centre for Adolescent and Adult Females with Congenital Abnormalities of the Genital Tract (UK)
- Mayer–Rokitansky–Küster–Hauser Syndrome on WebMD
| Classification |
|---|
