Multiple sulfatase deficiency
Multiple sulfatase deficiency (also known as "Austin disease",[1] and "mucosulfatidosis"[1]) is a very rare autosomal recessive[2]:561 lysosomal storage disease[3] caused by a deficiency in multiple sulfatase enzymes, or in formylglycine-generating enzyme, which activates sulfatases.[4]:502[5] It is similar to mucopolysaccharidosis.[6]
| Multiple sulfatase deficiency | |
|---|---|
| Other names | Juvenile sulfatidosis, Austin type |
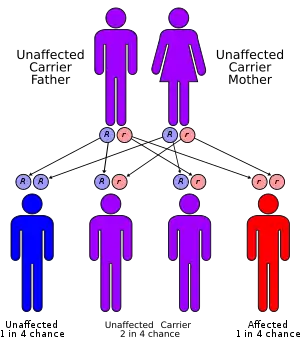 | |
| Multiple sulfatase deficiency is autorecessive | |
| Specialty | Endocrinology |
Signs and symptoms
Symptoms of this disorder commonly appear between one and two years of age. Symptoms include mildly coarsened facial features, deafness, ichthyosis[7] and an enlarged liver and spleen (hepatosplenomegaly).[8] Abnormalities of the skeleton, such as a curving of the spine and breast bone may occur. The skin of individuals afflicted with this disorder, is typically dry. Children affected by this disorder develop more slowly than normal and may display delayed speech and walking skills.
The disease is fatal, with symptoms that include neurological damage and severe mental retardation.[9] These sulfatase enzymes are responsible for breaking down and recycling complex sulfate-containing sugars from lipids and mucopolysaccharides within the lysosome. The accumulation of lipids and mucopolysaccharides inside the lysosome results in symptoms associated with this disorder. Worldwide, forty cases of Multiple Sulfatase Deficiency have been reported to date.
Causes
Multiple sulfatase deficiency is thought to be caused by any mutation of the SUMF1 gene which would render its protein product, the formylglycine-generating enzyme (FGE), defective.[10][11] These mutations result in inactive forms of FGE.[12] This enzyme is required for posttranslational modification of a cysteine residue in the sulfatase enzyme active site into formylglycine,[13] which is required for its proper function.[14]
Genetics
MSD has an autosomal recessive inheritance pattern.[2]:561 The inheritance probabilities per birth are as follows:
- If both parents are carriers:
- 25% (1 in 4) children will have the disorder
- 50% (2 in 4) children will be carriers (but unaffected)
- 25% (1 in 4) children will be free of MSD - unaffected child that is not a carrier
- If one parent is affected and one is free of MSD:
- 0% (0) children will have the disorder - only one parent is affected, other parent always gives normal gene
- 100% (4 in 4) children will be carriers (but unaffected)
- If one parent is a carrier and the other is free of MSD:
- 50% (2 in 4) children will be carriers (but unaffected)
- 50% (2 in 4) children will be free of MSD - unaffected child that is not a carrier
See also
- Linear porokeratosis
- List of cutaneous conditions
References
- Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- Dierks, T; Schmidt, B; Borissenko, Lv; Peng, J; Preusser, A; Mariappan, M; Von, Figura K (May 2003). "Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme". Cell. 113 (4): 435–44. doi:10.1016/S0092-8674(03)00347-7. PMID 12757705. S2CID 11571659.
- Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- Schmidt, B; Selmer, T; Ingendoh, A; Von, Figura K (July 1995). "A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency". Cell. 82 (2): 271–8. doi:10.1016/0092-8674(95)90314-3. PMID 7628016. S2CID 5864312.
- Soong BW, Casamassima AC, Fink JK, Constantopoulos G, Horwitz AL (1988). "Multiple sulfatase deficiency". Neurology. 38 (8): 1273–5. doi:10.1212/wnl.38.8.1273. PMID 2899861. S2CID 35222500.
- The American Heritage Medical Dictionary: mucosulfatidosis
- Burk, R; Valle, D; Thomas, GH; Miller, C; Moser, A; Moser, H; Rosenbaum, KN (1984). "Early manifestations of multiple sulfatase deficiency†". The Journal of Pediatrics. 104 (4): 574–8. doi:10.1016/S0022-3476(84)80550-8. PMID 6142938.
- Farooqui AA, Horrocks LA (1984). "Biochemical aspects of globoid and metachromatic leukodystrophies". Neurochem Pathol. 2 (3): 189–218. doi:10.1007/BF02834352. PMID 6152665. S2CID 36099212.
- Cosma MP, Pepe S, Annunziata I (May 2003). "The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases". Cell. 113 (4): 445–56. doi:10.1016/S0092-8674(03)00348-9. PMID 12757706. S2CID 15095377.
- Annunziata I, Bouchè V, Lombardi A (September 2007). "Multiple sulfatase deficiency is due to hypomorphic mutations of the SUMF1 gene". Human Mutation. 28 (9): 298. doi:10.1002/humu.9504. PMID 17657823. S2CID 8500605.
- Dierks T, Schmidt B, Borissenko LV (May 2003). "Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme". Cell. 113 (4): 435–44. doi:10.1016/S0092-8674(03)00347-7. PMID 12757705. S2CID 11571659.
- Preusser-Kunze A, Mariappan M, Schmidt B, Gande SL, Mutenda K, Wenzel D, von Figura K, Dierks T (April 2005). "Molecular Characterization of the Human C(alpha)-formylglycine-generating Enzyme". Journal of Biological Chemistry. 280 (15): 14900–14910. doi:10.1074/jbc.M413383200. PMID 15657036.
- Landgrebe J, Dierks T, Schmidt B (October 2003). "The human SUMF1 gene, required for posttranslational sulfatase modification, defines a new gene family which is conserved from pro- to eukaryotes". Gene. 316: 47–56. doi:10.1016/S0378-1119(03)00746-7. PMID 14563551.
External links
| Classification | |
|---|---|
| External resources |