Retinitis pigmentosa
Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision.[1] Symptoms include trouble seeing at night and decreased peripheral vision (side vision).[1] As peripheral vision worsens, people may experience "tunnel vision".[1] Complete blindness is uncommon.[2] Onset of symptoms is generally gradual and often in childhood.[1][2]
| Retinitis pigmentosa | |
|---|---|
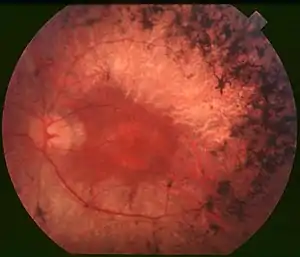 | |
| Back of the eye of a person with retinitis pigmentosa, mid stage. Note pigment deposits in the mid periphery along with retinal atrophy. While the macula is preserved there is some loss of pigmentation around it. | |
| Specialty | Ophthalmology |
| Symptoms | Trouble seeing at night, decrease peripheral vision[1] |
| Usual onset | Childhood[1] |
| Causes | Genetic[1] |
| Diagnostic method | Eye examination[1] |
| Treatment | Low vision aids, portable lighting, orientation and mobility training[1] |
| Medication | Vitamin A palmitate[1] |
| Frequency | 1 in 4,000 people[1] |
Retinitis pigmentosa is generally inherited from a person's parents.[1] Mutations in one of more than 50 genes are involved.[1] The underlying mechanism involves the progressive loss of rod photoreceptor cells in the back of the eye.[1] This is generally followed by loss of cone photoreceptor cells.[1] Diagnosis is by an examination of the retina finding dark pigment deposits.[1] Other supportive testing may include an electroretinogram, visual field testing, or genetic testing.[1]
There is currently no cure for retinitis pigmentosa.[2] Efforts to manage the problem may include the use of low vision aids, portable lighting, or orientation and mobility training.[1] Vitamin A palmitate supplements may be useful to slow worsening.[1] A visual prosthesis may be an option in certain people with severe disease.[1] It is estimated to affect 1 in 4,000 people.[1]
Signs and symptoms

The initial retinal degenerative symptoms of retinitis pigmentosa are characterized by decreased night vision (nyctalopia) and the loss of the mid-peripheral visual field.[3] The rod photoreceptor cells, which are responsible for low-light vision and are orientated in the retinal periphery, are the retinal processes affected first during non-syndromic forms of this disease.[4] Visual decline progresses relatively quickly to the far peripheral field, eventually extending into the central visual field as tunnel vision increases. Visual acuity and color vision can become compromised due to accompanying abnormalities in the cone photoreceptor cells, which are responsible for color vision, visual acuity, and sight in the central visual field.[4] The progression of disease symptoms occurs in a symmetrical manner, with both the left and right eyes experiencing symptoms at a similar rate.[5]
A variety of indirect symptoms characterize retinitis pigmentosa along with the direct effects of the initial rod photoreceptor degeneration and later cone photoreceptor decline. Phenomena such as photophobia, which describes the event in which light is perceived as an intense glare, and photopsia, the presence of blinking, swirling or shimmering lights within the visual field, often manifest during the later stages of RP. Findings related to RP have often been characterized in the fundus of the eye as the "ophthalamic triad". This includes the development of (1) a mottled appearance of the retinal pigment epithelium (RPE) caused by bony spicule formation, (2) a waxy appearance of the optic nerve, and (3) the attenuation of blood vessels in the retina.[3]
Non-syndromic RP usually presents a variety of the following symptoms:
- Night blindness
- Tunnel vision (due to loss of peripheral vision)
- Latticework vision
(due to loss of peripheral vision)
- loss of depth perception[6]
- Photopsia (blinking/swirling/shimmering lights)
- Photophobia (aversion to bright lights)
- Development of bone spicules in the fundus
- Slow adjustment from dark to light environments and vice versa
- Blurring of vision
- Poor color separation
- Loss of central vision
- Eventual blindness
Causes
RP may be: (1) Non-syndromic, that is, it occurs alone, without any other clinical findings, (2) Syndromic, with other neurosensory disorders, developmental abnormalities, or complex clinical findings, or (3) Secondary to other systemic diseases.[7]
- RP combined with deafness (congenital or progressive) is called Usher syndrome.[8]
- Alport's syndrome is associated with RP and an abnormal glomerular-basement membrane leading to nephrotic syndrome. It is inherited as X-linked dominant.
- RP combined with ophthalmoplegia, dysphagia, ataxia, and cardiac conduction defects is seen in the mitochondrial DNA disorder Kearns–Sayre syndrome (also known as Ragged Red Fiber Myopathy)
- RP combined with retardation, peripheral neuropathy, acanthotic (spiked) RBCs, ataxia, steatorrhea, and absence of VLDL is seen in abetalipoproteinemia.[9]
- RP is seen clinically in association with several other rare genetic disorders (including muscular dystrophy and chronic granulomatous disease) as part of McLeod syndrome. This is an X-linked recessive phenotype characterized by a complete absence of XK cell surface proteins, and therefore markedly reduced expression of all Kell red blood cell antigens. For transfusion purposes these patients are considered completely incompatible with all normal and K0/K0 donors.
- RP associated with hypogonadism, and developmental delay with an autosomal recessive inheritance pattern is seen with Bardet–Biedl syndrome[10]
Other conditions include neurosyphilis, toxoplasmosis and Refsum's disease.
Genetics
Retinitis pigmentosa (RP) is one of the most common forms of inherited retinal degeneration.[5]
There are multiple genes that, when mutated, can cause the retinitis pigmentosa phenotype.[11] Inheritance patterns of RP have been identified as autosomal dominant, autosomal recessive, X-linked, and maternally (mitochondrially) acquired, and are dependent on the specific RP gene mutations present in the parental generation.[12] In 1989, a mutation of the gene for rhodopsin, a pigment that plays an essential part in the visual transduction cascade enabling vision in low-light conditions, was identified. The rhodopsin gene encodes a principal protein of photoreceptor outer segments. Mutations in this gene most commonly presents as missense mutations or misfolding of the rhodopsin protein, and most frequently follow autosomal dominant inheritance patterns. Since the discovery of the rhodopsin gene, more than 100 RHO mutations have been identified, accounting for 15% of all types of retinal degeneration, and approximately 25% of autosomal dominant forms of RP.[5][13]
Up to 150 mutations have been reported to date in the opsin gene associated with the RP since the Pro23His mutation in the intradiscal domain of the protein was first reported in 1990. These mutations are found throughout the opsin gene and are distributed along the three domains of the protein (the intradiscal, transmembrane, and cytoplasmic domains). One of the main biochemical causes of RP in the case of rhodopsin mutations is protein misfolding, and the disruption of molecular chaperones.[14] It was found that the mutation of codon 23 in the rhodopsin gene, in which proline is changed to histidine, accounts for the largest fraction of rhodopsin mutations in the United States. Several other studies have reported various codon mutations associated with retinitis pigmentosa, including Thr58Arg, Pro347Leu, Pro347Ser, as well as deletion of Ile-255.[13][15][16][17][18] In 2000, a rare mutation in codon 23 was reported causing autosomal dominant retinitis pigmentosa, in which proline changed to alanine. However, this study showed that the retinal dystrophy associated with this mutation was characteristically mild in presentation and course. Furthermore, there was greater preservation in electroretinography amplitudes than the more prevalent Pro23His mutation.[19]
Autosomal recessive inheritance patterns of RP have been identified in at least 45 genes.[12] This means that two unaffected individuals who are carriers of the same RP-inducing gene mutation in diallelic form can produce offspring with the RP phenotype. A mutation on the USH2A gene is known to cause 10-15% of a syndromic form of RP known as Usher's Syndrome when inherited in an autosomal recessive fashion.[20]
Mutations in four pre-mRNA splicing factors are known to cause autosomal dominant retinitis pigmentosa. These are PRPF3 (human PRPF3 is HPRPF3; also PRP3), PRPF8, PRPF31 and PAP1. These factors are ubiquitously expressed and it is proposed that defects in a ubiquitous factor (a protein expressed everywhere) should only cause disease in the retina because the retinal photoreceptor cells have a far greater requirement for protein processing (rhodopsin) than any other cell type.[21]
The somatic, or X-linked inheritance patterns of RP are currently identified with the mutations of six genes, the most common occurring at specific loci in the RPGR and RP2 genes.[20]
Types include:
| OMIM | Gene | Type |
|---|---|---|
| 400004 | RPY | Retinitis pigmentosa Y-linked |
| 180100 | RP1 | Retinitis pigmentosa-1 |
| 312600 | RP2 | Retinitis pigmentosa-2 |
| 300029 | RPGR | Retinitis pigmentosa-3 |
| 608133 | PRPH2 | Retinitis pigmentosa-7 |
| 180104 | RP9 | Retinitis pigmentosa-9 |
| 180105 | IMPDH1 | Retinitis pigmentosa-10 |
| 600138 | PRPF31 | Retinitis pigmentosa-11 |
| 600105 | CRB1 | Retinitis pigmentosa-12, autosomal recessive |
| 600059 | PRPF8 | Retinitis pigmentosa-13 |
| 600132 | TULP1 | Retinitis pigmentosa-14 |
| 600852 | CA4 | Retinitis pigmentosa-17 |
| 601414 | HPRPF3 | Retinitis pigmentosa-18 |
| 601718 | ABCA4 | Retinitis pigmentosa-19 |
| 602772 | EYS | Retinitis pigmentosa-25 |
| 608380 | CERKL | Retinitis pigmentosa-26 |
| 607921 | FSCN2 | Retinitis pigmentosa-30 |
| 609923 | TOPORS | Retinitis pigmentosa-31 |
| 610359 | SNRNP200 | Retinitis pigmentosa 33 |
| 610282 | SEMA4A | Retinitis pigmentosa-35 |
| 610599 | PRCD | Retinitis pigmentosa-36 |
| 611131 | NR2E3 | Retinitis pigmentosa-37 |
| 268000 | MERTK | Retinitis pigmentosa-38 |
| 268000 | USH2A | Retinitis pigmentosa-39 |
| 612095 | PROM1 | Retinitis pigmentosa-41 |
| 612943 | KLHL7 | Retinitis pigmentosa-42 |
| 268000 | CNGB1 | Retinitis pigmentosa-45 |
| 613194 | BEST1 | Retinitis pigmentosa-50 |
| 613464 | TTC8 | Retinitis pigmentosa 51 |
| 613428 | C2orf71 | Retinitis pigmentosa 54 |
| 613575 | ARL6 | Retinitis pigmentosa 55 |
| 613617 | ZNF513 | Retinitis pigmentosa 58 |
| 613861 | DHDDS | Retinitis pigmentosa 59 |
| 613194 | BEST1 | Retinitis pigmentosa, concentric |
| 608133 | PRPH2 | Retinitis pigmentosa, digenic |
| 613341 | LRAT | Retinitis pigmentosa, juvenile |
| 268000 | SPATA7 | Retinitis pigmentosa, juvenile, autosomal recessive |
| 268000 | CRX | Retinitis pigmentosa, late-onset dominant |
| 300455 | RPGR | Retinitis pigmentosa, X-linked, and sinorespiratory infections, with or without deafness |
Pathophysiology

A variety of retinal molecular pathway defects have been matched to multiple known RP gene mutations. Mutations in the rhodopsin gene, which is responsible for the majority of autosomal-dominantly inherited RP cases, disrupts the rod-opsin protein essential for translating light into decipherable electrical signals within the phototransduction cascade of the central nervous system. Defects in the activity of this G-protein-coupled receptor are classified into distinct classes that depend on the specific folding abnormality and the resulting molecular pathway defects. The Class I mutant protein's activity is compromised as specific point mutations in the protein-coding amino acid sequence affect the pigment protein's transportation into the outer segment of the eye, where the phototransduction cascade is localized. Additionally, the misfolding of Class II rhodopsin gene mutations disrupts the protein's conjunction with 11-cis-retinal to induce proper chromophore formation. Additional mutants in this pigment-encoding gene affect protein stability, disrupt mRNA integrity post-translationally, and affect the activation rates of transducin and opsin optical proteins.[22]
Additionally, animal models suggest that the retinal pigment epithelium fails to phagocytose the outer rod segment discs that have been shed, leading to an accumulation of outer rod segment debris. In mice that are homozygous recessive for retinal degeneration mutation, rod photoreceptors stop developing and undergo degeneration before cellular maturation completes. A defect in cGMP-phosphodiesterase has also been documented; this leads to toxic levels of cGMP.
Diagnosis
An accurate diagnosis of retinitis pigmentosa relies on the documentation of the progressive loss photoreceptor cell function, confirmed by a combination of visual field and visual acuity tests, fundus and optical coherence imagery, and electroretinography (ERG).[23]
Visual field and acuity tests measure and compare the size of the patient's field of vision and the clarity of their visual perception with the standard visual measurements associated with healthy 20/20 vision. Clinical diagnostic features indicative of retinitis pigmentosa include a substantially small and progressively decreasing visual area in the visual field test, and compromised levels of clarity measured during the visual acuity test.[24] Additionally, optical tomography such as fundus and retinal (optical coherence) imagery provide further diagnostic tools when determining an RP diagnosis. Photographing the back of the dilated eye allows the confirmation of bone spicule accumulation in the fundus, which presents during the later stages of RP retinal degeneration. Combined with cross-sectional imagery of optical coherence tomography, which provides clues into photoreceptor thickness, retinal layer morphology, and retinal pigment epithelium physiology, fundus imagery can help determine the state of RP progression.[25]
While visual field and acuity test results combined with retinal imagery support the diagnosis of retinitis pigmentosa, additional testing is necessary to confirm other pathological features of this disease. Electroretinography (ERG) confirms the RP diagnosis by evaluating functional aspects associated with photoreceptor degeneration, and can detect physiological abnormalities before the initial manifestation of symptoms. An electrode lens is applied to the eye as photoreceptor response to varying degrees of quick light pulses is measured. Patients exhibiting the retinitis pigmentosa phenotype would show decreased or delayed electrical response in the rod photoreceptors, as well as possibly compromised cone photoreceptor cell response.
The patient's family history is also considered when determining a diagnosis due to the genetic mode of inheritance of retinitis pigmentosa. At least 35 different genes or loci are known to cause "nonsyndromic RP" (RP that is not the result of another disease or part of a wider syndrome). Indications of the RP mutation type can be determine through DNA testing, which is available on a clinical basis for:
- RLBP1 (autosomal recessive, Bothnia type RP)
- RP1 (autosomal dominant, RP1)
- RHO (autosomal dominant, RP4)
- RDS (autosomal dominant, RP7)
- PRPF8 (autosomal dominant, RP13)
- PRPF3 (autosomal dominant, RP18)
- CRB1 (autosomal recessive, RP12)
- ABCA4 (autosomal recessive, RP19)
- RPE65 (autosomal recessive, RP20)[26]
For all other genes (e.g. DHDDS), molecular genetic testing is available on a research basis only.
RP can be inherited in an autosomal dominant, autosomal recessive, X-linked or Y-linked[27] manner. X-linked RP can be either recessive, affecting primarily only males, or dominant, affecting both males and females, although males are usually more mildly affected. Some digenic (controlled by two genes) and mitochondrial forms have also been described.
Genetic counseling depends on an accurate diagnosis, determination of the mode of inheritance in each family, and results of molecular genetic testing.
Treatment
There is currently no cure for retinitis pigmentosa, but the efficacy and safety of various prospective treatments are currently being evaluated. The efficiency of various supplements, such as vitamin A, DHA, and lutein, in delaying disease progression remains an unresolved, yet prospective treatment option.[28][29] Clinical trials investigating optic prosthetic devices, gene therapy mechanisms, and retinal sheet transplantations are active areas of study in the partial restoration of vision in retinitis pigmentosa patients.[30]
Studies have demonstrated the delay of rod photoreceptor degeneration by the daily intake of 15000 IU (equivalent to 4.5 mg) of vitamin A palmitate; thus, stalling disease progression in some patients.[31] Recent investigations have shown that proper vitamin A supplementation can postpone blindness by up to 10 years (by reducing the 10% loss pa to 8.3% pa) in some patients in certain stages of the disease.[32]
The Argus retinal prosthesis became the first approved treatment for the disease in February 2011, and is currently available in Germany, France, Italy, and the UK.[33] Interim results on 30 patients long term trials were published in 2012.[34] The Argus II retinal implant has also received market approval in the US.[35] The device may help adults with RP who have lost the ability to perceive shapes and movement to be more mobile and to perform day-to-day activities. In June 2013, twelve hospitals in the US announced they would soon accept consultation for patients with RP in preparation for the launch of Argus II later that year.[36] The Alpha-IMS is a subretinal implant involving the surgical implantation of a small image-recording chip beneath the optic fovea. Measures of visual improvements from Alpha-IMS studies require the demonstration of the device's safety before proceeding with clinical trials and granting market approval.[37]
The goal of gene therapy studies is to virally supplement retinal cells expressing mutant genes associated with the retinitis pigmentosa phenotype with healthy forms of the gene; thus, allowing the repair and proper functioning of retinal photoreceptor cells in response to the instructions associated with the inserted healthy gene. Clinical trials investigating the insertion of the healthy RPE65 gene in retinas expressing the LCA2 retinitis pigmentosa phenotype measured modest improvements in vision; however, the degradation of retinal photoreceptors continued at the disease-related rate.[38] Likely, gene therapy may preserve remaining healthy retinal cells while failing to repair the earlier accumulation of damage in already diseased photoreceptor cells.[30] Response to gene therapy would theoretically benefit young patients exhibiting the shortest progression of photoreceptor decline; thus, correlating to a higher possibility of cell rescue via the healthy inserted gene.[39]
Prognosis
The progressive nature of and lack of a definitive cure for retinitis pigmentosa contribute to the inevitably discouraging outlook for patients with this disease. While complete blindness is rare, the person's visual acuity and visual field will continue to decline as initial rod photoreceptor and later cone photoreceptor degradation proceeds.[40]
Studies indicate that children carrying the disease genotype benefit from presymptomatic counseling in order to prepare for the physical and social implications associated with progressive vision loss. While the psychological prognosis can be slightly alleviated with active counseling[41] the physical implications and progression of the disease depend largely on the age of initial symptom manifestation and the rate of photoreceptor degradation, rather than access to prospective treatments. Corrective visual aids and personalized vision therapy provided by Low Vision Specialists may help patients correct slight disturbances in visual acuity and optimize their remaining visual field. Support groups, vision insurance, and lifestyle therapy are additional useful tools for those managing progressive visual decline.[23]
Epidemiology
Retinitis pigmentosa is the leading cause of inherited blindness,[42] with approximately 1/4,000 individuals experiencing the non-syndromic form of their disease within their lifetime.[43] It is estimated that 1.5 million people worldwide are currently affected. Early onset RP occurs within the first few years of life and is typically associated with syndromic disease forms, while late onset RP emerges from early to mid-adulthood.
Autosomal dominant and recessive forms of retinitis pigmentosa affect both male and female populations equally; however, the less frequent X-linked form of the disease affects male recipients of the X-linked mutation, while females usually remain unaffected carriers of the RP trait. The X-linked forms of the disease are considered severe, and typically lead to complete blindness during later stages. In rare occasions, a dominant form of the X-linked gene mutation will affect both males and females equally.[44]
Due to the genetic inheritance patterns of RP, many isolate populations exhibit higher disease frequencies or increased prevalence of a specific RP mutation. Pre-existing or emerging mutations that contribute to rod photoreceptor degeneration in retinitis pigmentosa are passed down through familial lines; thus, allowing certain RP cases to be concentrated to specific geographical regions with an ancestral history of the disease. Several hereditary studies have been performed to determine the varying prevalence rates in Maine (USA), Birmingham (England), Switzerland (affects 1/7000), Denmark (affects 1/2500), and Norway.[45] Navajo Indians display an elevated rate of RP inheritance as well, which is estimated as affecting 1 in 1878 individuals. Despite the increased frequency of RP within specific familial lines, the disease is considered non-discriminatory and tends to equally affect all world populations.
Research
Future treatments may involve retinal transplants,[46] artificial retinal implants,[47] gene therapy, stem cells, nutritional supplements, and/or drug therapies.
2012: Scientists at the University of Miami Bascom Palmer Eye Institute presented data showing protection of photoreceptors in an animal model when eyes were injected with mesencephalic astrocyte-derived neurotrophic factor (MANF).[48][49] Researchers at the University of California, Berkeley were able to restore vision to blind mice by exploiting a "photoswitch" that activates retinal ganglion cells in animals with damaged rod and cone cells.[50]
2015: A study by Bakondi et al. at Cedars-Sinai Medical Center showed that CRISPR/Cas9 can be used to treat rats with the autosomal dominant form of retinitis pigmentosa.[51] Researchers find that two molecules, rod-derived cone viability factor (RdCVF) and Nrf2, can protect cone photoreceptors in mouse models of retinitis pigmentosa.[52][53]
2016: RetroSense Therapeutics aimed to inject viruses with DNA from light-sensitive algae into the eyes of several blind people (who have retinitis pigmentosa). If successful, they will be able to see in black and white.[54][55]
In 2017 the FDA approved the gene therapy voretigene neparvovec to treat people with biallelic RPE65 mutation-associated retinal dystrophy.[56]
Notable cases
- Alex Bulmer, Canadian playwright[57]
- Molly Burke, Canadian YouTuber and motivational speaker.
- Neil Fachie, British paralympic cyclist[58]
- Lindy Hou, Australian tandem cyclist and triathlete[59]
- Steve Lonegan, Mayor of Bogota, New Jersey; Republican candidate for U.S. Senate[60]
- Amar Latif, entrepreneur, television personality and professional traveller.
- Akbar Khan, musician & disability activist from India.
- Woody Shaw, American jazz trumpeter.[61]
- Shel Talmy, American record producer, songwriter and arranger[62]
- Danelle Umstead, American Paralympic alpine skier, Dancing with the Stars (U.S. TV series) contestant[63]
- Jon Wellner, American actor[64]
- Steve Wynn, American business magnate and Las Vegas casino developer[65]
See also
References
- "Facts About Retinitis Pigmentosa". National Eye Institute. May 2014. Retrieved 18 April 2020.
- Openshaw, Amanda (Feb 2008). Understanding Retinitis Pigmentosa (PDF). University of Michigan Kellogg Eye Center. Archived from the original (PDF) on 2017-08-29. Retrieved 2017-12-02.
- Shintani, Kelly; Shechtman, Diana L.; Gurwood, Andrew S. (2009). "Review and update: Current treatment trends for patients with retinitis pigmentosa". Optometry. 80 (7): 384–401. doi:10.1016/j.optm.2008.01.026. PMID 19545852.
- Soucy, E; Wang, Y; Nirenberg, S; Nathans, J; Meister, M (1998). "A Novel Signaling Pathway from Rod Photoreceptors to Ganglion Cells in Mammalian Retina". Neuron. 21 (3): 481–93. doi:10.1016/S0896-6273(00)80560-7. PMID 9768836. S2CID 6636037.
- Hartong, Dyonne T; Berson, Eliot L; Dryja, Thaddeus P (2006). "Retinitis pigmentosa". The Lancet. 368 (9549): 1795–1809. doi:10.1016/S0140-6736(06)69740-7. PMID 17113430. S2CID 24950783.
- Prem Senthil, M; Khadka, J; Pesudovs, K (May 2017). "Seeing through their eyes: lived experiences of people with retinitis pigmentosa". Eye. 31 (5): 741–748. doi:10.1038/eye.2016.315. PMC 5437327. PMID 28085147.
- Daiger, S P; Sullivan, L S; Bowne, S J (2013). "Genes and mutations causing retinitis pigmentosa". Clinical Genetics. 84 (2): 132–41. doi:10.1111/cge.12203. PMC 3856531. PMID 23701314.
- "Usher Syndrome".
- "Diseases – MM – Types Of Overview". Muscular Dystrophy Association. 2015-12-18.
- "Bardet-Biedl (Laurence Moon)".
- Online Mendelian Inheritance in Man (OMIM): RETINITIS PIGMENTOSA; RP - 268000
- Rivolta, C.; Sharon, D; Deangelis, M. M.; Dryja, T. P. (2002). "Retinitis pigmentosa and allied diseases: Numerous diseases, genes, and inheritance patterns". Human Molecular Genetics. 11 (10): 1219–27. doi:10.1093/hmg/11.10.1219. PMID 12015282.
- Berson, Eliot L.; Rosner, B; Sandberg, M. A.; Dryja, T. P. (1991). "Ocular Findings in Patients with Autosomal Dominant Retinitis Pigmentosa and a Rhodopsin Gene Defect (Pro-23-His)". Archives of Ophthalmology. 109 (1): 92–101. doi:10.1001/archopht.1991.01080010094039. PMID 1987956.
- Senin, Ivan I.; Bosch, Laia; Ramon, Eva; Zernii, Evgeni Yu.; Manyosa, Joan; Philippov, Pavel P.; Garriga, Pere (2006). "Ca2+/recoverin dependent regulation of phosphorylation of the rhodopsin mutant R135L associated with retinitis pigmentosa". Biochemical and Biophysical Research Communications. 349 (1): 345–52. doi:10.1016/j.bbrc.2006.08.048. PMID 16934219.
- Dryja, Thaddeus P.; McGee, Terri L.; Reichel, Elias; Hahn, Lauri B.; Cowley, Glenn S.; Yandell, David W.; Sandberg, Michael A.; Berson, Eliot L. (1990). "A point mutation of the rhodopsin gene in one form of retinitis pigmentosa". Nature. 343 (6256): 364–6. Bibcode:1990Natur.343..364D. doi:10.1038/343364a0. PMID 2137202. S2CID 4351328.
- Dryja, Thaddeus P.; McGee, Terri L.; Hahn, Lauri B.; Cowley, Glenn S.; Olsson, Jane E.; Reichel, Elias; Sandberg, Michael A.; Berson, Eliot L. (1990). "Mutations within the Rhodopsin Gene in Patients with Autosomal Dominant Retinitis Pigmentosa". New England Journal of Medicine. 323 (19): 1302–7. doi:10.1056/NEJM199011083231903. PMID 2215617.
- Berson, E. L.; Rosner, B; Sandberg, M. A.; Weigel-Difranco, C; Dryja, T. P. (1991). "Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine". American Journal of Ophthalmology. 111 (5): 614–23. doi:10.1016/s0002-9394(14)73708-0. PMID 2021172.
- Inglehearn, C. F.; Bashir, R; Lester, D. H.; Jay, M; Bird, A. C.; Bhattacharya, S. S. (1991). "A 3-bp deletion in the rhodopsin gene in a family with autosomal dominant retinitis pigmentosa". American Journal of Human Genetics. 48 (1): 26–30. PMC 1682750. PMID 1985460.
- Oh, Kean T.; Weleber, R. G.; Lotery, A; Oh, D. M.; Billingslea, A. M.; Stone, E. M. (2000). "Description of a New Mutation in Rhodopsin, Pro23Ala, and Comparison with Electroretinographic and Clinical Characteristics of the Pro23His Mutation". Archives of Ophthalmology. 118 (9): 1269–76. doi:10.1001/archopht.118.9.1269. PMID 10980774.
- "Retinitis pigmentosa".
- Bujakowska, K.; Maubaret, C.; Chakarova, C. F.; Tanimoto, N.; Beck, S. C.; Fahl, E.; Humphries, M. M.; Kenna, P. F.; Makarov, E.; Makarova, O.; Paquet-Durand, F.; Ekstrom, P. A.; Van Veen, T.; Leveillard, T.; Humphries, P.; Seeliger, M. W.; Bhattacharya, S. S. (2009). "Study of Gene-Targeted Mouse Models of Splicing Factor Gene Prpf31 Implicated in Human Autosomal Dominant Retinitis Pigmentosa (RP)". Investigative Ophthalmology & Visual Science. 50 (12): 5927–5933. doi:10.1167/iovs.08-3275. PMID 19578015.
- Mendes HF, van der Spuy J, Chapple JP, Cheetham ME (April 2005). "Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy". Trends in Molecular Medicine. 11 (4): 177–185. doi:10.1016/j.molmed.2005.02.007. PMID 15823756.
- "Understanding Retinitis Pigmentosa" (PDF). Archived from the original (PDF) on 2017-03-29. Retrieved 2015-03-16.
- Abigail T Fahim (1993). "Nonsyndromic Retinitis Pigmentosa Overview". Retinitis Pigmentosa Overview. University of Washington, Seattle.
- Chang S, Vaccarella L, Olatunji S, Cebulla C, Christoforidis J (2011). "Diagnostic Challenges in Retinitis Pigmentosa: Genotypic Multiplicity and Phenotypic Variability". Current Genomics. 12 (4): 267–75. doi:10.2174/138920211795860116. PMC 3131734. PMID 22131872.
- "Retinitis Pigmentosa".
- Zhao GY, Hu DN, Xia HX, Xia ZC (1995). "Chinese family with retinitis pigmentosa". Ophthalmic Genetics. 16 (2): 75–76. doi:10.3109/13816819509056916. PMID 7493160.
- Hartong, Dyonne T; Berson, Eliot L; Dryja, Thaddeus P (November 2006). "Retinitis pigmentosa". The Lancet. 368 (9549): 1795–1809. doi:10.1016/S0140-6736(06)69740-7. PMID 17113430. S2CID 24950783.
- Schwartz, Stephen G; Wang, Xue; Chavis, Pamela; Kuriyan, Ajay E; Abariga, Samuel A (18 June 2020). "Vitamin A and fish oils for preventing the progression of retinitis pigmentosa". Cochrane Database of Systematic Reviews. 6: CD008428. doi:10.1002/14651858.CD008428.pub3. PMC 7388842. PMID 32573764.
- Lok, Corie (September 2014). "Curing blindness: Vision quest". Nature. 513 (7517): 160–162. Bibcode:2014Natur.513..160L. doi:10.1038/513160a. PMID 25209781.
- Berson, Eliot L.; Rosner, B; Sandberg, M. A.; Hayes, K. C.; Nicholson, B. W.; Weigel-Difranco, C; Willett, W (1993). "A Randomized Trial of Vitamin a and Vitamin E Supplementation for Retinitis Pigmentosa". Archives of Ophthalmology. 111 (6): 761–72. doi:10.1001/archopht.1993.01090060049022. PMID 8512476.
- Berson, Eliot L. (2007). "Long-term visual prognoses in patients with retinitis pigmentosa: The Ludwig von Sallmann lecture". Experimental Eye Research. 85 (1): 7–14. doi:10.1016/j.exer.2007.03.001. PMC 2892386. PMID 17531222.
- "Nahrungsergänzungsmittel: ALLES, was du wissen musst!". Archived from the original on 2013-08-19. Retrieved 2013-08-19.
- Humayun, Mark S.; Dorn, Jessy D.; Da Cruz, Lyndon; Dagnelie, Gislin; Sahel, José-Alain; Stanga, Paulo E.; Cideciyan, Artur V.; Duncan, Jacque L.; Eliott, Dean; Filley, Eugene; Ho, Allen C.; Santos, Arturo; Safran, Avinoam B.; Arditi, Aries; Del Priore, Lucian V.; Greenberg, Robert J.; Argus Ii Study, Group (2012). "Interim Results from the International Trial of Second Sight's Visual Prosthesis". Ophthalmology. 119 (4): 779–88. doi:10.1016/j.ophtha.2011.09.028. PMC 3319859. PMID 22244176.
- https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm339824.htm%5B%5D
- "'First Bionic Eye' Retinal Chip for Blind". Science Daily. 29 June 2013. Retrieved 30 June 2013.
- Stingl K, Bartz-Schmidt KU, Besch D, Braun A, Bruckmann A, Gekeler F, Greppmaier U, Hipp S, Hörtdörfer G, Kernstock C, Koitschev A, Kusnyerik A, Sachs H, Schatz A, Stingl KT, Peters T, Wilhelm B, Zrenner E (2013). "Artificial vision with wirelessly powered subretinal electronic implant alpha-IMS". Proc. Biol. Sci. 280 (1757): 20130077. doi:10.1098/rspb.2013.0077. PMC 3619489. PMID 23427175.
- Bainbridge, James W.B.; Smith, Alexander J.; Barker, Susie S.; Robbie, Scott; Henderson, Robert; Balaggan, Kamaljit; Viswanathan, Ananth; Holder, Graham E.; Stockman, Andrew; Tyler, Nick; Petersen-Jones, Simon; Bhattacharya, Shomi S.; Thrasher, Adrian J.; Fitzke, Fred W.; Carter, Barrie J.; Rubin, Gary S.; Moore, Anthony T.; Ali, Robin R. (2008). "Effect of Gene Therapy on Visual Function in Leber's Congenital Amaurosis". New England Journal of Medicine. 358 (21): 2231–9. CiteSeerX 10.1.1.574.4003. doi:10.1056/NEJMoa0802268. PMID 18441371.
- Maguire AM, High KA, Auricchio A, et al. (November 2009). "Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial". The Lancet. 374 (9701): 1597–1605. doi:10.1016/S0140-6736(09)61836-5. PMC 4492302. PMID 19854499.
- Shintani, Kelly; Shechtman, Diana L.; Gurwood, Andrew S. (July 2009). "Review and update: Current treatment trends for patients with retinitis pigmentosa". Optometry - Journal of the American Optometric Association. 80 (7): 384–401. doi:10.1016/j.optm.2008.01.026. PMID 19545852.
- Mezer, E; Babul-Hirji, R; Wise, R; Chipman, M; Dasilva, L; Rowell, M; Thackray, R; Shuman, C. T.; Levin, A. V. (2007). "Attitudes regarding predictive testing for retinitis pigmentosa". Ophthalmic Genet. 28 (1): 9–15. doi:10.1080/13816810701199423. PMID 17454742. S2CID 21636488.
- Parmeggiani F (2011). "Clinics, Epidemiology and Genetics of Retinitis Pigmentosa". Current Genomics. 12 (4): 236–7. doi:10.2174/138920211795860080. PMC 3131730. PMID 22131868.
- Hamel, Christian (2006). "Retinitis pigmentosa". Orphanet Journal of Rare Diseases. 1: 40. doi:10.1186/1750-1172-1-40. PMC 1621055. PMID 17032466.
- Prokisch, Holger; Hartig, Monika; Hellinger, Rosa; Meitinger, Thomas; Rosenberg, Thomas (2007). "IOVS – A Population-Based Epidemiological and Genetic Study of X-Linked Retinitis Pigmentosa". Investigative Ophthalmology & Visual Science. 48 (9): 4012–8. doi:10.1167/iovs.07-0071. PMID 17724181.
- Haim, Marianne (2002). "The epidemiology of retinitis pigmentosa in Denmark". Acta Ophthalmologica Scandinavica. 80 (233): 1–34. doi:10.1046/j.1395-3907.2002.00001.x. PMID 11921605.
- Graham-Rowe, Duncan (8 September 2008). "Retinal transplants see fleeting success". Nature: news.2008.1088. doi:10.1038/news.2008.1088.
- "Ophthalmologists Implant Five Patients with Artificial Silicon Retina Microchip To Treat Vision Loss from Retinitis Pigmentosa" (Press release). Rush University Medical Center. 2005-01-31. Archived from the original on 2005-02-08. Retrieved 2007-06-16.
- Wen, Rong; Luo, Lingyu; Huang, Dequang; Xia, Xin; Wang, Zhengying; Chen, Pingping; Li, Yiwen (March 2012). "Mesencephalic Astrocyte-derived Neurotrophic Factor (MANF) Protects Rod and Cone Photoreceptors from Degeneration in Transgenic Rats Carrying the S334ter Rhodopsin Mutation". Invest. Ophthalmol. Vis. Sci. 53 (14): 2581. Retrieved 2016-08-07.
- Wen, Rong; Luo, Lingyu; Huang, Dequang; Xia, Xin; Wang, Zhengying; Chen, Pingping; Li, Yiwen (May 7, 2012). Mesencephalic Astrocyte-derived Neurotrophic Factor (MANF) Protects Rod and Cone Photoreceptors from Degeneration in Transgenic Rats Carrying the S334ter Rhodopsin Mutation. ARVO 2012.
- Tochitsky, Ivan; Polosukhina, Aleksandra; Degtyar, Vadim E.; Gallerani, Nicholas; Smith, Caleb M.; Friedman, Aaron; Van Gelder, Russell N.; Trauner, Dirk; Kaufer, Daniela; Kramer, Richard H. (2014). "Restoring Visual Function to Blind Mice with a Photoswitch that Exploits Electrophysiological Remodeling of Retinal Ganglion Cells". Neuron. 81 (4): 800–13. doi:10.1016/j.neuron.2014.01.003. PMC 3933823. PMID 24559673.
- Bakondi, Benjamin; Lv, Wenjian; Lu, Bin; Jones, Melissa K; Tsai, Yuchun; Kim, Kevin J; Levy, Rachelle; Akhtar, Aslam Abbasi; Breunig, Joshua J; Svendsen, Clive N; Wang, Shaomei (March 2016). "In Vivo CRISPR/Cas9 Gene Editing Corrects Retinal Dystrophy in the S334ter-3 Rat Model of Autosomal Dominant Retinitis Pigmentosa". Molecular Therapy. 24 (3): 556–563. doi:10.1038/mt.2015.220. PMC 4786918. PMID 26666451. Lay summary.
- Byrne, Leah C.; Dalkara, Deniz; Luna, Gabriel; Fisher, Steven K.; Clérin, Emmanuelle; Sahel, Jose-Alain; Léveillard, Thierry; Flannery, John G. (2 January 2015). "Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration". Journal of Clinical Investigation. 125 (1): 105–116. doi:10.1172/JCI65654. PMC 4382269. PMID 25415434.
- Xiong, Wenjun; MacColl Garfinkel, Alexandra E.; Li, Yiqing; Benowitz, Larry I.; Cepko, Constance L. (1 April 2015). "NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage". Journal of Clinical Investigation. 125 (4): 1433–1445. doi:10.1172/JCI79735. PMC 4396467. PMID 25798616.
- "FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss". U.S. Food and Drug Administration. 19 December 2017. Retrieved 18 June 2020.
- Bourzac, Katherine. "A blind woman in Texas is first person to undergo optogenetic therapy, which could let her see again if successful". technologyreview.com.
- Commissioner, Office of the (2018-11-03). "Press Announcements - FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss". www.fda.gov. Retrieved 2019-01-16.
- Maga, Carly (12 December 2017). "Blind actor Alex Bulmer leads the way into theatre's future". Toronto Star. Retrieved 9 August 2020.
- Neil Fachie http://www.paralympics.org.uk/gb/athletes/neil-fachie%5B%5D
- McDonald, Margie (31 May 2008). "Wheel turns a full circle as proud Lindy rides for two countries in Beijing". The Australian. p. 54. Retrieved 1 February 2012.
- Rizzo, Salvador (2013-09-25). "Lonegan opens up about is blindness".
- Spencer, Frederick J. (2002). Jazz and Death: Medical Profiles of Jazz Greats. University of Mississippi Press. pp. 55–57. ISBN 9781578064533.
- Wayne, Artie. "SHEL TALMY INTERVIEWED BY ARTIE WAYNE, PART TWO". spectropop.com. Artie Wayne. Retrieved 31 March 2020.
- "Danelle Umstead". Team USA. Retrieved 2018-09-13.
- "CSI Cast: Jon Wellner". CBS. Retrieved October 5, 2010.
- Paumgarten, Nick (2006-10-16). "Doh! Dept: The $40-Million Elbow". The New Yorker. Retrieved 2012-08-13.
External links
| Classification | |
|---|---|
| External resources |