Nemaline myopathy
Nemaline myopathy (also called rod myopathy or nemaline rod myopathy) is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births.[1] It is the most common non-dystrophic myopathy.[2][3]
| Nemaline myopathy | |
|---|---|
| Other names | Nemaline rod myopathy |
| Specialty | Neurology |
"Myopathy" means muscle disease. Muscle fibers from a person with nemaline myopathy contains thread-like[4] rods, sometimes called nemaline bodies.[5] While the rods are diagnostic of the disorder, they are more likely a byproduct of the disease process rather than causing any dysfunction on their own. People with nemaline myopathy (NM) usually experience delayed motor development, or no motor development in severe cases, and weakness may occur in all of the skeletal muscles, such as muscles in the arms, legs, torso, neck flexors, throat, and face. The weakness tends to be more severe in the proximal muscles rather than the distal muscles. The ocular muscles are normally spared.
The disorder is often clinically categorized into groups with wide ranges of overlapping severity, from the most severe neonatal form which is incompatible with life, to a form so mild that it may not be diagnosed since the person appears to function at the lowest end of normal strength and breathing adequacy. Sporadic late onset nemaline myopathy (SLONM) is not a congenital disorder and is considered a different muscle disease from NM, which has its onset at birth or early childhood.[6] Respiratory problems are usually a primary concern for people with all forms of NM, and respiratory infections are quite common. NM shortens life expectancy, particularly in the more severe forms, but aggressive and proactive care allows most individuals to survive and even lead active lives.
Nemaline myopathy is one of the neuromuscular diseases covered by the Muscular Dystrophy Association in the United States.
Signs and symptoms
Signs vary from person to person. Young children and babies lack movement and have a difficult time eating and breathing. For young children not diagnosed immediately at birth, these are usually the first visible symptoms. One sign is a swollen face in disproportional areas. Other examples in newborns include swaying and a difficulty in moving. Other symptoms include feeble muscles in the neck and upper rib cage area. In adults, the most common symptom is respiratory problems.[7] Other symptoms in adults could range from mild to severe speech impediments. It is common to be diagnosed with scoliosis in relations to nemaline myopathy.[8] As babies that have NM develop and become of age when they should start walking, many take longer than average due to the lack of muscle, or just muscle fatigue.[9]
Since facial muscles are involved in NM takeover, elongated faces and a lower mandible are often observed in people with NM. People affected by NM usually will begin to feel muscle exhaustion between ages 20–50. NM is usually not progressive. Gastroesophageal reflux, although not common, is associated with NM. Heart abnormalities can occur as a result of NM, but the likelihood of that happening are not high.[7]
Mobility and orthopedics
Most children with mild NM eventually walk independently, although often at a later age than their peers. Some use wheelchairs or other devices, such as walkers or braces, to enhance their mobility. Individuals with severe NM generally have limited limb movement and use wheelchairs full-time. Due to weakness in the trunk muscles, people with NM are prone to scoliosis, which usually develops in childhood and worsens during puberty. Many individuals with NM undergo spinal fusion surgery to straighten and stabilize their backs.
Although patients early on often have mobility in their joints that is past the normal range, as they age, joint deformities and scoliosis usually occur.[10] If the person with nemaline myopathy keeps an eye on his or her joints early on, the problems with them can be detected when they begin and their progression can be delayed. Treatment of joint problems ranges from stretching exercises with physical therapy to surgical introduction of braces. The benefits of exercise in people with nemaline myopathy are still being studied, however, researchers have seen improvements in muscle function from low-intensity exercise. Vigorous exercise and the use of heavy weights should be avoided.
Respiratory involvement
Attention to respiratory issues is critical to the health of many people with NM. Infants with severe NM frequently experience respiratory distress at or soon after birth, although this is only found in the rarest forms. Though respiratory compromise may not be immediately apparent in people with intermediate or mild NM, it often, but not always, exists to some extent. As in many neuromuscular disorders, hypoventilation can begin insidiously, and it may cause serious health problems if not remedied by the use of noninvasive mechanical devices to assist breathing, particularly at night.
Communication and eating
Bulbar (throat) muscle weakness is a main feature of nemaline myopathy. Individuals with the most severe forms of NM are unable to swallow and receive their nutrition through feeding tubes. Most people with intermediate and mild NM take some or all of their nutrition orally. Bulbar muscle impairment may also lead to difficulty with communication. People with NM often have hypernasal speech as a result of poor closure of the velopharyngeal port (between the soft palate and the back of the throat). This may be able to be surgically corrected. Communicative skills may be enhanced through speech therapy, oral prosthetic devices, surgery, and augmentative communication devices. NM does not have any impact on cognition or intelligence.
Physical characteristics and effects
Physical expression of nemaline myopathy varies greatly, but weakness is usually concentrated in the proximal muscles, particularly respiratory, bulbar and trunk muscles.[11] People with severe NM show obvious symptoms at birth, while those with intermediate or mild NM may initially appear unaffected. Babies with NM are frequently observed to be "floppy" and hypotonic. Children born with NM often gain strength as they grow, though the effect of muscle weakness on body features may become more evident with time. Adults with NM typically have a very slender physique.[11]
Causes
Nemaline myopathy is caused by mutations in one of at least 11 different genes.[2][12] Nemaline myopathy is a clinically and genetically heterogeneous disorder and both autosomal dominant and autosomal recessive forms can occur. Diagnosis is made based upon clinical signs such as muscle weakness, absent or low deep tendon reflexes (hyporeflexia), and a high-arched palate, along with electron-dense aggregates, called nemaline rods, being observed at the microscopic level within muscle fibers. Genetic confirmation through identification of a known genetic mutation in the patient is also an important component of diagnosis.[13][14]
The two most common gene mutations causing nemaline myopathy are found on NEB or ACTA1.[15] Mutations of the NEB gene usually result in symptoms present at birth or beginning in early childhood. This mutation results in about 50% of affected nemaline myopathy patients. The most common inheritance pathway for those with mutations in NEB is autosomal recessive in which each parent carries one mutated copy along with one normal functioning copy of the gene, and they pass the mutated copy to their offspring. In some cases, occasionally with ACTA1 mutations, NM can be caused by an inheritance pattern of autosomal dominance. This mutation results in about 15 to 25 percent of NM cases.[16] One reason why this is lower is because NM is associated with de novo mutations in ACTA1, occurring spontaneously in the egg or sperm.[9] When the condition is heritable, each pregnancy with the same partners has the same risk of passing the mutated genes to offspring. New mutations (de novo) can also occur causing NM and de novo mutations have been most often found to occur in the ACTA1 gene.[16] MYPN is the last found gene related to NM The risk of all cases of nemaline myopathy is the same in males and females.[9]

| Gene 1 | Proportion of Nemaline Myopathy Attributed to Mutation of This Gene | Test Method |
|---|---|---|
| NEB | Up to 50% | Sequence analysis |
| Deletion/duplication analysis & targeted analysis for pathogenic variants | ||
| ACTA1 | 15%-25% | Sequence analysis |
| Deletion/duplication analysis | ||
| TPM3 | 2%-3% | Sequence analysis |
| Deletion/duplication analysis | ||
| TPM2 | <1% | Sequence analysis |
| Deletion/duplication analysis | ||
| TNNT1 | Almost exclusively in Old Amish | Sequence analysis |
| Deletion/duplication analysis | ||
| CFL2 | Rare | Sequence analysis |
| KBTBD13 | Unknown | Sequence analysis |
| KLHL40 | 20% | Sequence analysis |
| KLHL41 | Unknown | Sequence analysis |
| LMOD3 | Unknown | Sequence analysis |
| Unknown | NA | NA |
The physical capabilities of a given person with NM do not correlate well either with genotype or with muscle pathology as observed in the biopsy.[18]
Mechanism
Muscle cells contract in complex mechanical and chemical processes. If any part of the process or structure is disrupted, dysfunction will likely result, as in the case of those with genetic variations. In those with nemaline myopathy, muscle contraction is adversely affected. At the electron microscopic level, rod-shaped components can often be seen in some of the muscle cells, and when seen, are diagnostic for the condition called nemaline rod myopathy. The presence of these rods is not itself causing muscle weakness; rather they appear as a result of something going wrong within the muscle fiber. There is no connection between the number of rods found in the muscle cells and the amount of weakness a person has. All of the different gene mutations leading to the condition called nemaline myopathy that have been found so far are in genes that encode different components of the sarcomere.[11] In normal muscle cells, the various parts of the muscle fibers that make up the sarcomere are distributed evenly in a pattern for effective muscle contraction. Evidence suggests that some kinds of NM affect the arrangement of these muscle fibers, causing the muscles to be unable to contract as efficiently or effectively.
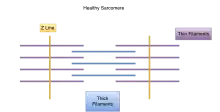
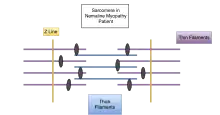
Nemaline myopathy is usually genetic and shows traits in the affected individual from birth or an early age. However, there are some cases of symptoms of nemaline myopathy not showing up until adulthood. These cases are usually not genetic. Of the genes that have been linked to nemaline myopathy, most are also involved in encoding proteins in the sarcomeres in the muscle cells.[11] Respiratory muscles are often more affected than other skeletal muscle groups. Cardiac muscle is usually not affected in nemaline myopathy; however, in cases where it does, patients often present with dilated cardiomyopathy. The ocular muscles are usually spared.
The different genes whose mutations lead to the different kinds of nemaline myopathies affect the cells and the person's body differently. The first kind of nemaline myopathy identified is due to the Slow α-Tropomyosin Gene TPM3 and varies from case to case with its severity. In this kind of nemaline myopathy, affected people are weaker and more affected in their lower limbs than their upper limbs.[11]
As stated above, the most common genetic form of NM is caused by a mutation in the nebulin gene, called Nebulin,[19][20] and has a range of severity levels. All published cases up to this point where NM is thought to be caused by a mutation in the NEB gene have been autosomal recessive and are the most common cause of nemaline myopathy.[21] Patients with this kind of NM are more affected in the muscles in their head, rather than their proximal muscles at the core of their body. Consequently, patients with this genetic mutation often cannot lift their heads and speak with a nasal voice. There have been cases that suggest this kind of NM may lead to patients having higher intellect.[11]
A third kind of nemaline myopathy in the Skeletal Muscle α-Actin Gene ACTA1 is due to a recessive null mutation.[11] These patients do not always show the typical nemaline bodies in their muscle cells. The only abnormality they show is an abnormal distribution of muscle fibers.
There are several other identified kinds of mutations that lead to Nemaline Myopathies. One affects slow skeletal muscles, one leads to the formation of both nemaline bodies and other abnormal, core-like, structures forming in the patient's muscles.
Diagnosis
- Electromyography or (EMG). This procedure determines if nerve or muscle cells are damaged. Since a common symptom of Nemaline Myopathy is muscle weakness this allows doctors to determine where and why the weakness is occurring.[7]
- MRI of the Musculoskeletal System. MRI uses a magnetic field to take pictures of body structures and allows physicians to determine if a patient has a certain disease.[7]
- Needle biopsy A needle biopsy allows a physician to test specific cells in the body. These cells are sent to a laboratory to undergo testing and can further determine why muscle weakness throughout the body could be occurring. This testing can confirm that muscle cells contain rod like structures.[7]
Treatment
At present, Nemaline myopathy does not have a cure. Nemaline myopathy is a very rare disease that only affects 1 out of 50,000 on average, although recent studies show that this number is even smaller. There are a number of treatments to minimize the symptoms of the disease. The treatments and procedures to help patients with nemaline myopathy vary depending on the severity of the disease. A possible accommodation could be the use of a stabilizer, such as a brace. Other means include moderate stretching and moderate exercise to help target muscles maintain maximum health.[22] As people with NM grow and develop throughout their lives, it is important for them to see a variety of health professionals regularly, including a neurologist, physical therapist, and others, such as speech therapists and psychologists, to help both the patient and family adjust to everyday life.[10]
Outcome
Although there is no cure for NM, it is possible, and common for many people live healthy active lives even with moderate to severe cases.[23] Research continues to seek ways to ameliorate debilitating symptoms and lengthen the life-span in quality ways for those affected. Some people have seen mild improvements in secretion handling, energy level, and physical functioning with supplemental L-tyrosine, an amino acid that is available through health centers.[24] Some symptoms may worsen as the patient ages. Muscle loss increases with age naturally, but it is even more significant with nemaline myopathy.[25]
Current research
New research resources have become available for the NM community, such as the CMDIR (registry) and the CMD-TR (biorepository). These two resources connect families and individuals interested in participating in research with the scientists that aim to treat or cure NM. Some research on NM seeks to better understand the molecular effects the gene mutations have on muscle cells and the rest of the body[26] and to observe any connections NM may have to other diseases and health complications.
History
"Rod myopathy" was first identified by Douglas Reye, an Australian physician, in 1958.[27] However, Reye's results were never published because another doctor dismissed his finding of rods in the muscle tissue as an artifact of the biopsy. Forty years later, Reye's "rod myopathy" patient was confirmed to have nemaline myopathy. Another group of Australian researchers has since published an article recognizing Reye for his work.[28]
"Nemaline myopathy" was first named in a published paper in 1963 by North American researchers P.E. Cohen and G. M. Shy. Shy and his team discovered rod- like structures in muscle fibers of patients with muscle weakness by performing muscle biopsies on multiple patients.[22] Laboratories performing research on NM are located around the world, notably in the United States, Canada, England, Finland, and Australia.
Society
In 1999, the first website on nemaline myopathy was launched, and in October 2004, the first Nemaline Myopathy Convention was held in Toronto, Canada. Many more conferences and social events have been held since, and all events organized since 2008 have been co-sponsored by A Foundation Building Strength for Nemaline Myopathy (AFBS),[29] the only foundation focused on supporting treatment development and social events for the NM community. In March 2006, Niki Shisler released a book, Fragile, in which she recounted her experiences surrounding the birth of twin sons with severe NM. In 2014, a team of experts collaborated with affected individuals and families caring for someone with a congenital myopathy to develop the first guidebook on managing life with a congenital myopathy
References
- Yuen, Michaela; Sandaradura, Sarah A.; Dowling, James J.; Kostyukova, Alla S.; Moroz, Natalia; Quinlan, Kate G.; Lehtokari, Vilma-Lotta; Ravenscroft, Gianina; Todd, Emily J. (2014-11-03). "Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy". The Journal of Clinical Investigation. 124 (11): 4693–4708. doi:10.1172/JCI75199. ISSN 0021-9738. PMC 4347224. PMID 25250574.
- de Winter, Josine Marieke; Buck, Danielle; Hidalgo, Carlos; Jasper, Jeffrey R.; Malik, Fady I.; Clarke, Nigel F.; Stienen, Ger J. M.; Lawlor, Michael W.; Beggs, Alan H. (2013-06-01). "Troponin activator augments muscle force in nemaline myopathy patients with nebulin mutations". Journal of Medical Genetics. 50 (6): 383–392. doi:10.1136/jmedgenet-2012-101470. ISSN 1468-6244. PMC 3865762. PMID 23572184.
- Ottenheijm, Coen A. C.; Lawlor, Michael W.; Stienen, Ger J. M.; Granzier, Henk; Beggs, Alan H. (2011-05-15). "Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy". Human Molecular Genetics. 20 (10): 2015–2025. doi:10.1093/hmg/ddr084. ISSN 1460-2083. PMC 3080611. PMID 21357678.
- nemaline myopathy
- "Nemaline Myopathy - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Retrieved 2016-04-25.
- Chahin, Nizar; Selcen, Duygu; Engel, Andrew G. (2005-10-25). "Sporadic late onset nemaline myopathy". Neurology. 65 (8): 1158–1164. doi:10.1212/01.wnl.0000180362.90078.dc. ISSN 1526-632X. PMID 16148261. S2CID 23334154.
- "Muscle Disorders: MedlinePlus". www.nlm.nih.gov. Retrieved 2016-04-10.
- "Diseases - Inherited / Endocrine Myopathies - Type Of (Nemaline myopathy)". Muscular Dystrophy Association. 2015-12-18. Retrieved 2016-04-10.
- Piteau, SJ; Rossiter, JP; Smith, RG; MacKenzie, JJ (2014). "Congenital Myopathy With Cap-Like Structures and Nemaline Rods: Case Report and Literature Review". Pediatric Neurology. Elsevier. 51 (2): 192–197. doi:10.1016/j.pediatrneurol.2014.04.002. PMID 25079567.
- Ohlsson, Monica. "Nemaline Myopathy". Socialstyrelsen. The Swedish Information Centre for Rare Diseases.
- Wallgren, Petterson; Sewry, CA; Nowak, KJ; Laing, NG (2011). "Nemaline Myopathies". Semin Pediatr Neurol. 18 (4): 230–8. doi:10.1016/j.spen.2011.10.004. PMID 22172418.
- Sandaradura, Sarah A.; North, Kathryn N. (2015-08-26). "LMOD3: the "missing link" in nemaline myopathy?". Oncotarget. 6 (29): 26548–26549. doi:10.18632/oncotarget.5267. ISSN 1949-2553. PMC 4694930. PMID 26337340.
- North, Kathryn N.; Wang, Ching H.; Clarke, Nigel; Jungbluth, Heinz; Vainzof, Mariz; Dowling, James J.; Amburgey, Kimberly; Quijano-Roy, Susana; Beggs, Alan H. (2014-02-01). "Approach to the diagnosis of congenital myopathies". Neuromuscular Disorders. 24 (2): 97–116. doi:10.1016/j.nmd.2013.11.003. ISSN 1873-2364. PMC 5257342. PMID 24456932.
- Romero, Norma B.; Sandaradura, Sarah A.; Clarke, Nigel F. (2013-10-01). "Recent advances in nemaline myopathy". Current Opinion in Neurology. 26 (5): 519–526. doi:10.1097/WCO.0b013e328364d681. ISSN 1473-6551. PMID 23995272. S2CID 12185842.
- Wallgren-Pettersson, Carina; Sewry, Caroline A.; Nowak, Kristen J.; Laing, Nigel G. (2011-12-01). "Nemaline myopathies". Seminars in Pediatric Neurology. 18 (4): 230–238. doi:10.1016/j.spen.2011.10.004. ISSN 1558-0776. PMID 22172418.
- Laing, Nigel G.; Dye, Danielle E.; Wallgren-Pettersson, Carina; Richard, Gabriele; Monnier, Nicole; Lillis, Suzanne; Winder, Thomas L.; Lochmüller, Hanns; Graziano, Claudio (2009-09-01). "Mutations and Polymorphisms of the Skeletal Muscle α-Actin Gene (ACTA1)". Human Mutation. 30 (9): 1267–1277. doi:10.1002/humu.21059. ISSN 1059-7794. PMC 2784950. PMID 19562689.
- North, Kathryn N.; Ryan, Monique M. (2015-06-11). "Table 1. [Summary of Molecular Genetic Testing Used in Nemaline Myopathy]. - GeneReviews® - NCBI Bookshelf". Retrieved 2016-04-25.
- Ryan MM, Ilkovski B, Strickland CD, et al. (February 2003). "Clinical course correlates poorly with muscle pathology in nemaline myopathy". Neurology. 60 (4): 665–73. doi:10.1212/01.WNL.0000046585.81304.BC. PMID 12601110. S2CID 11652174.
- "WikiGenes - Collaborative Publishing". WikiGenes - Collaborative Publishing. Retrieved 2016-04-10.
- Li, Frank; Buck, Danielle; De Winter, Josine; Kolb, Justin; Meng, Hui; Birch, Camille; Slater, Rebecca; Escobar, Yael Natelie; Smith, John E. (2015-09-15). "Nebulin deficiency in adult muscle causes sarcomere defects and muscle-type-dependent changes in trophicity: novel insights in nemaline myopathy". Human Molecular Genetics. 24 (18): 5219–5233. doi:10.1093/hmg/ddv243. ISSN 1460-2083. PMC 4550825. PMID 26123491.
- Ottenheijm, Lawlor, Steiner, Granzier, Beggs. "Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy"
- "Nemaline Myopathy - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Retrieved 2016-04-10.
- "Diseases-Inherited/Endocrine Myopathies". Muscular Dystrophy Association. 2015-12-18. Retrieved 27 April 2016.
- Ryan, Monique M.; Sy, Catherine; Rudge, Sian; Ellaway, Carolyn; Ketteridge, David; Roddick, Laurence G.; Iannaccone, Susan T.; Kornberg, Andrew J.; North, Kathryn N. (2008-06-01). "Dietary L-tyrosine supplementation in nemaline myopathy". Journal of Child Neurology. 23 (6): 609–613. doi:10.1177/0883073807309794. ISSN 0883-0738. PMID 18079309. S2CID 23371464.
- "Nemaline myopathy". www.socialstyrelsen.se. Retrieved 2016-04-10.
- "Nemaline Myopathy". Genetics Home Reference. U.S. National Library of Medicine.
- "Congenital myopathies and muscular dystrophies team". Westmead Children's Hospital. 9 August 2005. Retrieved 13 February 2012.
- Schnell C, Kan A, North KN (June 2000). "'An artefact gone awry': identification of the first case of nemaline myopathy by Dr R.D.K. Reye". Neuromuscul. Disord. 10 (4–5): 307–12. doi:10.1016/S0960-8966(99)00123-6. PMID 10838259. S2CID 38543084.
- "A Foundation Building Strength for Nemaline Myopathy Research".
External links
| Classification | |
|---|---|
| External resources |
| Types | |
|---|---|
| National/International Organizations | |
| National/International Events |
|
| Clinical trials | |