Pharmacodynamics
Pharmacodynamics (PD) is the study of the biochemical and physiologic effects of drugs (especially pharmaceutical drugs). The effects can include those manifested within animals (including humans), microorganisms, or combinations of organisms (for example, infection).
Pharmacodynamics and pharmacokinetics are the main branches of pharmacology, being itself a topic of biology interested in the study of the interactions between both endogenous and exogenous chemical substances with living organisms.
In particular, pharmacodynamics is the study of how a drug affects an organism, whereas pharmacokinetics is the study of how the organism affects the drug. Both together influence dosing, benefit, and adverse effects. Pharmacodynamics is sometimes abbreviated as PD and pharmacokinetics as PK, especially in combined reference (for example, when speaking of PK/PD models).
Pharmacodynamics places particular emphasis on dose–response relationships, that is, the relationships between drug concentration and effect.[1] One dominant example is drug-receptor interactions as modeled by

where L, R, and LR represent ligand (drug), receptor, and ligand-receptor complex concentrations, respectively. This equation represents a simplified model of reaction dynamics that can be studied mathematically through tools such as free energy maps.
Effects on the body
The majority of drugs either
- mimic or inhibit normal physiological/biochemical processes or inhibit pathological processes in animals or
- inhibit vital processes of endo- or ectoparasites and microbial organisms.
There are 7 main drug actions:[3]
- stimulating action through direct receptor agonism and downstream effects
- depressing action through direct receptor agonism and downstream effects (ex.: inverse agonist)
- blocking/antagonizing action (as with silent antagonists), the drug binds the receptor but does not activate it
- stabilizing action, the drug seems to act neither as a stimulant or as a depressant (ex.: some drugs possess receptor activity that allows them to stabilize general receptor activation, like buprenorphine in opioid dependent individuals or aripiprazole in schizophrenia, all depending on the dose and the recipient)
- exchanging/replacing substances or accumulating them to form a reserve (ex.: glycogen storage)
- direct beneficial chemical reaction as in free radical scavenging
- direct harmful chemical reaction which might result in damage or destruction of the cells, through induced toxic or lethal damage (cytotoxicity or irritation)
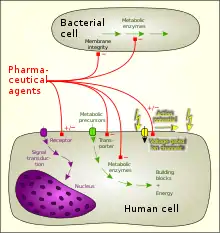
Desired activity
The desired activity of a drug is mainly due to successful targeting of one of the following:
- Cellular membrane disruption
- Chemical reaction with downstream effects
- Interaction with enzyme proteins
- Interaction with structural proteins
- Interaction with carrier proteins
- Interaction with ion channels
- Ligand binding to receptors:
- Hormone receptors
- Neuromodulator receptors
- Neurotransmitter receptors
General anesthetics were once thought to work by disordering the neural membranes, thereby altering the Na+ influx. Antacids and chelating agents combine chemically in the body. Enzyme-substrate binding is a way to alter the production or metabolism of key endogenous chemicals, for example aspirin irreversibly inhibits the enzyme prostaglandin synthetase (cyclooxygenase) thereby preventing inflammatory response. Colchicine, a drug for gout, interferes with the function of the structural protein tubulin, while Digitalis, a drug still used in heart failure, inhibits the activity of the carrier molecule, Na-K-ATPase pump. The widest class of drugs act as ligands that bind to receptors that determine cellular effects. Upon drug binding, receptors can elicit their normal action (agonist), blocked action (antagonist), or even action opposite to normal (inverse agonist).
In principle, a pharmacologist would aim for a target plasma concentration of the drug for a desired level of response. In reality, there are many factors affecting this goal. Pharmacokinetic factors determine peak concentrations, and concentrations cannot be maintained with absolute consistency because of metabolic breakdown and excretory clearance. Genetic factors may exist which would alter metabolism or drug action itself, and a patient's immediate status may also affect indicated dosage.
Undesirable effects
Undesirable effects of a drug include:
- Increased probability of cell mutation (carcinogenic activity)
- A multitude of simultaneous assorted actions which may be deleterious
- Interaction (additive, multiplicative, or metabolic)
- Induced physiological damage, or abnormal chronic conditions
Therapeutic window
The therapeutic window is the amount of a medication between the amount that gives an effect (effective dose) and the amount that gives more adverse effects than desired effects. For instance, medication with a small pharmaceutical window must be administered with care and control, e.g. by frequently measuring blood concentration of the drug, since it easily loses effects or gives adverse effects.
Duration of action
The duration of action of a drug is the length of time that particular drug is effective.[4] Duration of action is a function of several parameters including plasma half-life, the time to equilibrate between plasma and target compartments, and the off rate of the drug from its biological target.[5]
Receptor binding and effect
The binding of ligands (drug) to receptors is governed by the law of mass action which relates the large-scale status to the rate of numerous molecular processes. The rates of formation and un-formation can be used to determine the equilibrium concentration of bound receptors. The equilibrium dissociation constant is defined by:

where L=ligand, R=receptor, square brackets [] denote concentration. The fraction of bound receptors is

Where is the fraction of receptor bound by the ligand.

This expression is one way to consider the effect of a drug, in which the response is related to the fraction of bound receptors (see: Hill equation). The fraction of bound receptors is known as occupancy. The relationship between occupancy and pharmacological response is usually non-linear. This explains the so-called receptor reserve phenomenon i.e. the concentration producing 50% occupancy is typically higher than the concentration producing 50% of maximum response. More precisely, receptor reserve refers to a phenomenon whereby stimulation of only a fraction of the whole receptor population apparently elicits the maximal effect achievable in a particular tissue.
The simplest interpretation of receptor reserve is that it is a model that states there are excess receptors on the cell surface than what is necessary for full effect. Taking a more sophisticated approach, receptor reserve is an integrative measure of the response-inducing capacity of an agonist (in some receptor models it is termed intrinsic efficacy or intrinsic activity) and of the signal amplification capacity of the corresponding receptor (and its downstream signaling pathways). Thus, the existence (and magnitude) of receptor reserve depends on the agonist (efficacy), tissue (signal amplification ability) and measured effect (pathways activated to cause signal amplification). As receptor reserve is very sensitive to agonist's intrinsic efficacy, it is usually defined only for full (high-efficacy) agonists.[6][7][8]
Often the response is determined as a function of log[L] to consider many orders of magnitude of concentration. However, there is no biological or physical theory that relates effects to the log of concentration. It is just convenient for graphing purposes. It is useful to note that 50% of the receptors are bound when [L]=Kd .
The graph shown represents the conc-response for two hypothetical receptor agonists, plotted in a semi-log fashion. The curve toward the left represents a higher potency (potency arrow does not indicate direction of increase) since lower concentrations are needed for a given response. The effect increases as a function of concentration.
Multicellular pharmacodynamics
The concept of pharmacodynamics has been expanded to include Multicellular Pharmacodynamics (MCPD). MCPD is the study of the static and dynamic properties and relationships between a set of drugs and a dynamic and diverse multicellular four-dimensional organization. It is the study of the workings of a drug on a minimal multicellular system (mMCS), both in vivo and in silico. Networked Multicellular Pharmacodynamics (Net-MCPD) further extends the concept of MCPD to model regulatory genomic networks together with signal transduction pathways, as part of a complex of interacting components in the cell.[9]
Toxicodynamics
Pharmacokinetics and pharmacodynamics are termed toxicokinetics and toxicodynamics in the field of ecotoxicology. Here, the focus is on toxic effects on a wide range of organisms. The corresponding models are called toxicokinetic-toxicodynamic models.[10]
See also
References
- Lees P, Cunningham FM, Elliott J (2004). "Principles of pharmacodynamics and their applications in veterinary pharmacology". J. Vet. Pharmacol. Ther. 27 (6): 397–414. doi:10.1111/j.1365-2885.2004.00620.x. PMID 15601436.
- Duffus, J. (1 January 1993). "Glossary for chemists of terms used in toxicology (IUPAC Recommendations 1993)". Pure and Applied Chemistry. 65 (9): 2003–2122. doi:10.1351/pac199365092003.
- "Introduction to Pharmacology". PsychDB.
- Carruthers SG (February 1980). "Duration of drug action". Am. Fam. Physician. 21 (2): 119–26. PMID 7352385.
- Vauquelin G, Charlton SJ (October 2010). "Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action". Br. J. Pharmacol. 161 (3): 488–508. doi:10.1111/j.1476-5381.2010.00936.x. PMC 2990149. PMID 20880390.
- Ruffolo RR Jr (December 1982). "Review important concepts of receptor theory". J. Auton. Pharmacol. 2 (4): 277–295. doi:10.1111/j.1474-8673.1982.tb00520.x. PMID 7161296.
- Dhalla AK, Shryock JC, Shreeniwas R, Belardinelli L (2003). "Pharmacology and therapeutic applications of A1 adenosine receptor ligands". Curr. Top. Med. Chem. 3 (4): 369–385. doi:10.2174/1568026033392246. PMID 12570756.
- Gesztelyi R, Kiss Z, Wachal Z, Juhasz B, Bombicz M, Csepanyi E, Pak K, Zsuga J, Papp C, Galajda Z, Branzaniuc K, Porszasz R, Szentmiklosi AJ, Tosaki A (2013). "The surmountable effect of FSCPX, an irreversible A(1) adenosine receptor antagonist, on the negative inotropic action of A(1) adenosine receptor full agonists in isolated guinea pig left atria". Arch. Pharm. Res. 36 (3): 293–305. doi:10.1007/s12272-013-0056-z. PMID 23456693. S2CID 13439779.
- Zhao, Shan; Iyengar, Ravi (2012). "Systems Pharmacology: Network Analysis to Identify Multiscale Mechanisms of Drug Action". Annual Review of Pharmacology and Toxicology. 52: 505–521. doi:10.1146/annurev-pharmtox-010611-134520. ISSN 0362-1642. PMC 3619403. PMID 22235860.
- Li Q, Hickman M (2011). "Toxicokinetic and toxicodynamic (TK/TD) evaluation to determine and predict the neurotoxicity of artemisinins". Toxicology. 279 (1–3): 1–9. doi:10.1016/j.tox.2010.09.005. PMID 20863871.
External links
| Wikimedia Commons has media related to Pharmacodynamics. |
- Vijay. (2003) Predictive software for drug design and development. Pharmaceutical Development and Regulation 1 ((3)), 159–168.
- Werner, E., In silico multicellular systems biology and minimal genomes, DDT vol 8, no 24, pp 1121–1127, Dec 2003. (Introduces the concepts MCPD and Net-MCPD)
- Dr. David W. A. Bourne, OU College of Pharmacy Pharmacokinetic and Pharmacodynamic Resources.
- Introduction to Pharmacokinetics and Pharmacodynamics