Retosiban
Retosiban also known as GSK-221,149-A[1][2] is an oral drug which acts as an oxytocin receptor antagonist. It is being developed by GlaxoSmithKline for the treatment of preterm labour.[3][4] Retosiban has high affinity for the oxytocin receptor (Ki = 0.65 nM) and has greater than 1400-fold selectivity[5] over the related vasopressin receptors
 | |
| Clinical data | |
|---|---|
| Other names | GSK-221149-A |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C27H34N4O5 |
| Molar mass | 494.592 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
Mechanism of action
Retosiban is a competitive oxytocin receptor antagonist which blocks the oxytocin-mediated contraction of the uterine smooth muscle in the female uterus that occurs during the initiation of preterm labour. This has been used to prevent preterm labour and premature birth.
Pharmacology
Retosiban has been shown to be an effective tocolytic. By intravenous and oral administration it produces a dose-dependent decrease in oxytocin-induced uterine contractions in non-pregnant female rats. In late-term pregnant rats it significantly reduces spontaneous uterine contractions in a dose-dependent manner by intravenous administration.[5] In humans retosiban prolongs pregnancy and reduces preterm birth. Intravenous administration of retosiban in women with spontaneous preterm labour was associated with a greater than 1-week increase in time to delivery compared with placebo, a significant reduction in preterm deliveries, a non-significant increase in uterine quiescence, and a favourable safety profile. The results demonstrate proof-of-concept in the treatment of threatened spontaneous preterm labour [6]
Pharmacokinetics
The oral bioavailability of retosiban is in the order of 100% in the rat with a half life of 1.4 hours. It has low to moderate intrinsic clearance in microsomes from three pre-clinical species (rat, dog, cynomolgus monkey) and low intrinsic clearance in human microsomes. It has a good cytochrome P450 (Cyp450) profile with no significant inhibition, with IC50 > 100μM, low protein binding (<80%) and low predicted CNS penetration.[4]
Physical and chemical properties
At physiological pH, retosiban exists in an uncharged state. It has good solubility (> 0.22 mg/ml), with a logd of 2.2.[4]
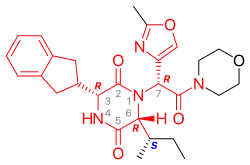
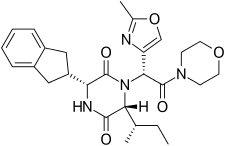
Retosiban consists of a central 2,5 diketopiperazine ring with an R-indanyl group at the 3 position and an R (S-secButyl) at the 6 position, both cis to each other, and with a R-2-methyl oxazole ring at the 7 position in the acyclic amide attached to the N1-position. Retosiban is the (3R, 6R, 7R)-isomer and is a sub-nanomolar (Ki = 0.65 nM) oxytocin receptor antagonist, while the (3R, 6R, 7S)-isomer where the stereochemistry in the amide side-chain at C-7 is inverted, is 10-fold less potent. Typically in this series of 2,5 diketopiperazine oxytocin antagonists the (3S, 6S, 7S) isomer is >500 less active than the (3R, 6R, 7R)-isomer. In addition to the 2,5 diketopiperazine essential core, retosiban also contains several structural characteristics that improve its effectiveness and safety. An indanyl group at position 3 is the best choice in terms of oxytocin receptor antagonist potency, its replacement by phenethyl and benzyl groups led to a progressive weakening of activity. At C-3, a 4-carbon branched alkyl was shown to be preferred with R (S-secButyl) being the best; smaller alkyl groups result in reduced antagonist activity.[4] The 2-methyl oxazole ring at the 7 position gives good aqueous solubility, low protein binding and minimal Cyp450 interaction. This structure–activity relationship (SAR) is supported by the crystal structure of the human oxytocin receptor in complex with retosiban, [7] where the lipophilic indanyl substituent penetrates into a deep, mainly hydrophobic crevice at the bottom of the binding pocket, while the oxazol-morpholine amide moiety is closest to the extracellular surface. The oxazole ring is the most solvent-exposed substituent, and the morpholine ring has no direct interactions with the receptor. The 2,5-diketopiperazine core specifically interacts with the receptor through a polar interaction interface.
Synthesis
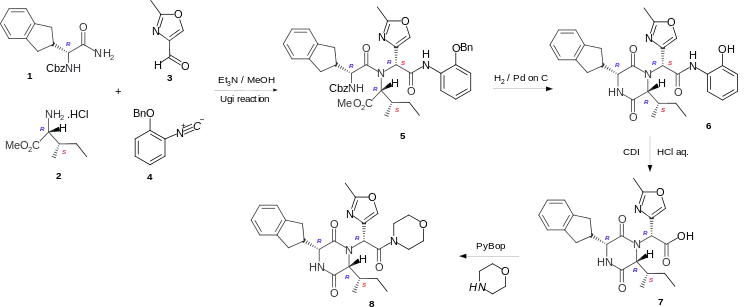
Retosiban is a cyclic dipeptide or 2,5-diketopiperazine and these are formed by cyclising the corresponding linear dipeptide. In the short lab-scale and highly stereoselective synthesis of Retosiban 8 the linear peptide 5 is formed by the four-component Ugi reaction of the carboxybenzyl (Cbz) protected R-indanylglycine 1, D-alloisoleucine methyl ester hydrochloride 2, 2-methyloxazole-4-carboxaldehyde 3 and 2-benzyloxyphenylisonitrile 4. Hydrogenation to remove the Cbz and benzyl protecting groups, enabled cyclization of the linear peptide 5 to occur to give the phenolic cyclic dipeptide 6. Hydrolysis of the phenolic amide, by reaction with carbonyl diimidazole (CDI), followed addition of aqueous hydrochloric acid gave the acid 7 which was converted to the amide Retosiban 8 by activating the acid with the peptide coupling reagent PyBOP (benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate) followed by the addition of morpholine.[4] Although the linear peptide 5 and the cyclic dipeptide 6 are a mixture of diastereoisomers (7RS) at the exocyclic amide, the hydrochloric acid hydrolysis of the activated phenolic amide caused epimerisation at the exocyclic position and yielded the acid 7 with the required (7R)-stereochemistry as the major product.
References
- Liddle J, Allen MJ, Borthwick AD, Brooks DP, Davies DE, Edwards RM, et al. (January 2008). "The discovery of GSK221149A: a potent and selective oxytocin antagonist". Bioorganic & Medicinal Chemistry Letters. 18 (1): 90–4. doi:10.1016/j.bmcl.2007.11.008. PMID 18032036.
- Borthwick AD, Liddle J (January 2013). "Retosiban and Epelsiban: Potent and Selective Orally available Oxytocin Antagonists". In Domling A (ed.). Methods and Principles in Medicinal Chemistry: Protein-Protein Interactions in Drug Discovery. Weinheim: Wiley-VCH. pp. 225–256. doi:10.1002/9783527648207.ch10. ISBN 978-3-527-33107-9.
- USAN Council (2007). "Statement on a Nonproprietary Name Adopted by the USAN Council" (PDF).
- Borthwick AD, Liddle J (July 2011). "The Design of Orally Bioavailable 2,5-Diketopiperazine Oxytocin Antagonists: From Concept to Clinical Candidate for Premature Labour". Medicinal Research Reviews. 31 (4): 576–604. doi:10.1002/med.20193. PMID 20027670.
- McCafferty GP, Pullen MA, Wu C, Edwards RM, Allen MJ, Woollard PM, Borthwick AD, Liddle J, Hickey DM, Brooks DP, Westfall TD (2007). "Use of a novel and highly selective oxytocin receptor antagonist to characterize uterine contractions in the rat". American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 293 (1): R299–305. doi:10.1152/ajpregu.00057.2007. PMID 17395790.
- Thornton S, Miller H, Valenzuela G, Snidow J, Stier B, Fossler MJ, Montague TH, Powell M, Beach KJ (October 2015). "Treatment of spontaneous preterm labour with retosiban: a phase 2 proof‐of‐concept study". British Journal of Clinical Pharmacology. 80 (4): 740–749. doi:10.1111/bcp.12646. PMC 4594710. PMID 25819462.
- Waltenspühl Y, Schöppe J, Ehrenmann J, Kummer L, Plückthun A (July 2020). "Crystal structure of the human oxytocin receptor". Science Advances. 6 (29): 1–11. doi:10.1126/sciadv.abb5419. PMID 32832646.
